A/Prof. Ken Rodgers School of Life Sciences
Learning Objectives
- Explain how drug clearance (CL) determines the steady-state plasma concentration
- Calculate the half-life, volume of distribution, clearance and loading doses of drugs
- Explain how the timecourse of drug clearance can be described using one or two compartment kinetic models
- Understand differences between first-order and zero-order (saturation) kinetics
- Explain the pharmacokinetics of alcohol, including why alcohol has a constant rate of metabolism per hour
References
- Rang HP, Dale MM, Ritter JM, Flower R and Henderson G (2015) Pharmacology, 8th Edition, Churchill Livingstone, Sydney.
- Drug metabolism and elimination – Chapter 10
Definitions
Definitions
Pharmacokinetics = Time course of drug concentrations attained in different compartments in the body during or after dosing
Time course of drug concentration in plasma or blood

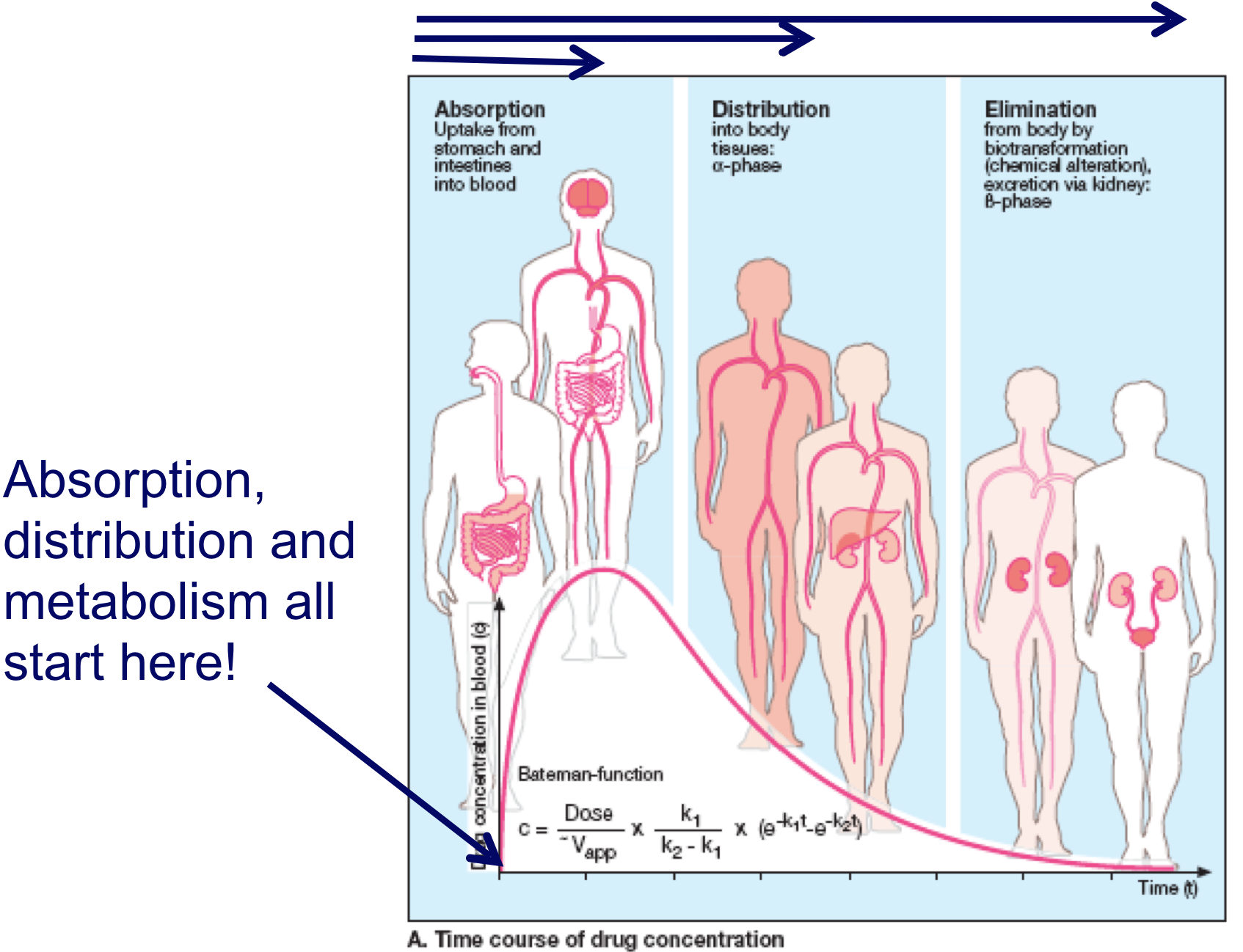
Time course of drug concentration
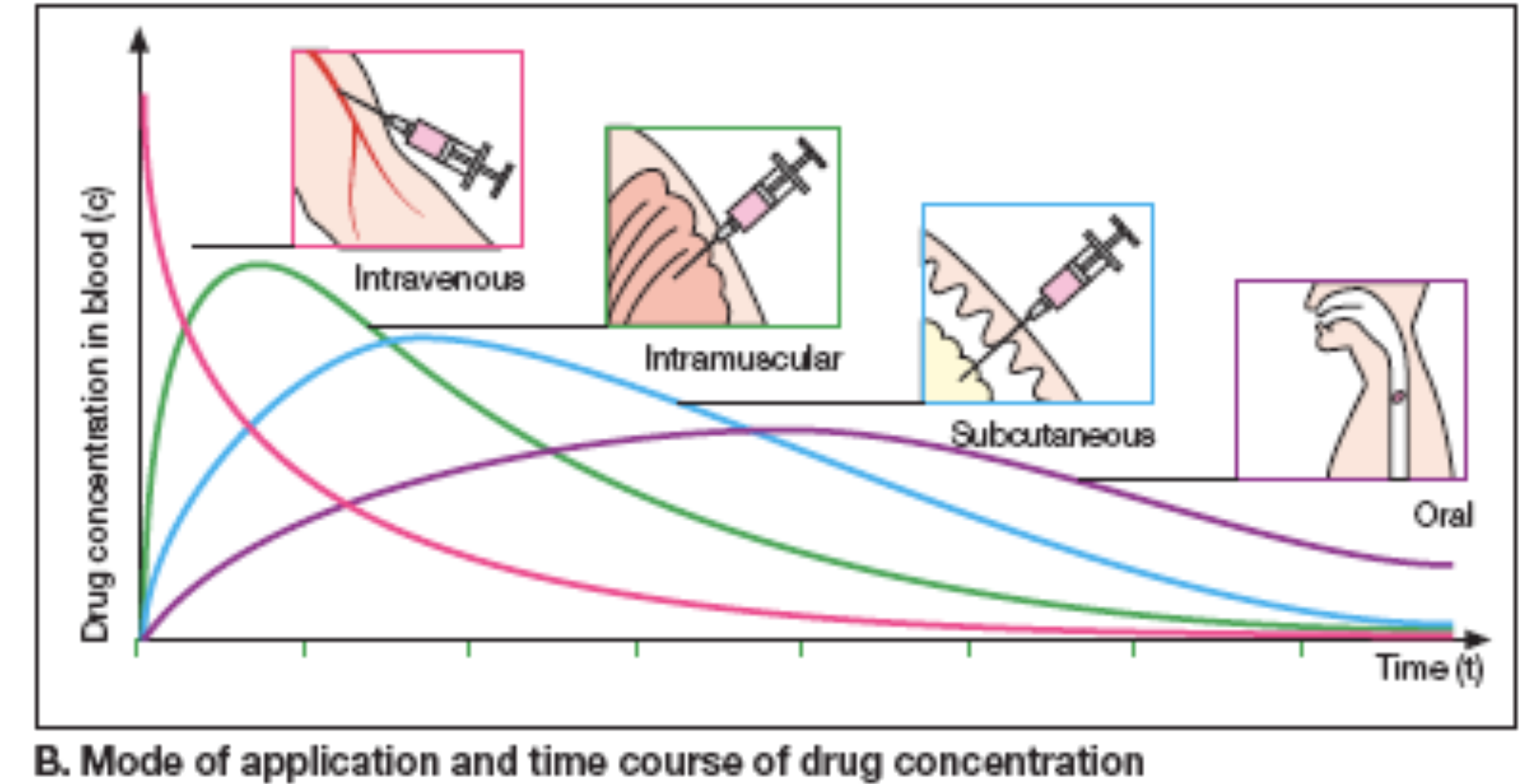
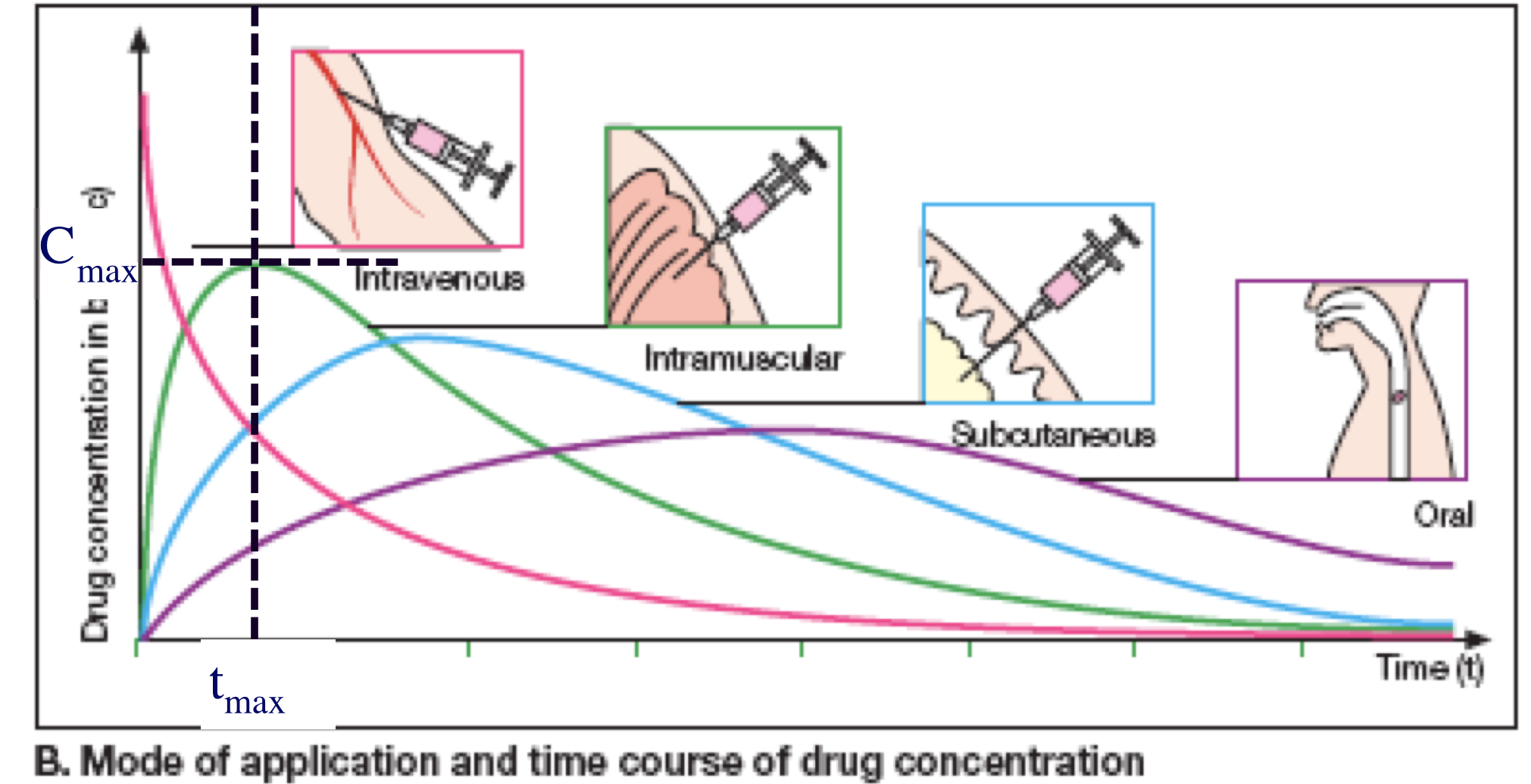
Drug Elimination
- The irreversible loss of drug from the body. This can occur via:
- Metabolism
- Excretion


Drug clearance 1
- Clearance (CL) is the volume of plasma cleared of drug per unit time (L/hr)
- Relates rate of drug elimination to its plasma conc (Cp) (mg/L)
- Rate of drug elimination = Cp x CL (mg/hr)
- For most individuals at therapeutic concs CL is the same at different drug doses (Q) (NB: not in overdose)
Drug clearance
Check out animation on http://sepia.unil.ch/pharmacology/index.php?id=62
(This is the best visual representation of drug clearance I could find)
Drug Clearance 2
Clearance
- describes the efficiency of elimination
- is additive – so is made up of a number of components
CLtotal = CLrenal + CLnon-renal
CLtotal = CLrenal+ CLmetab + CLbile + CLlung +….
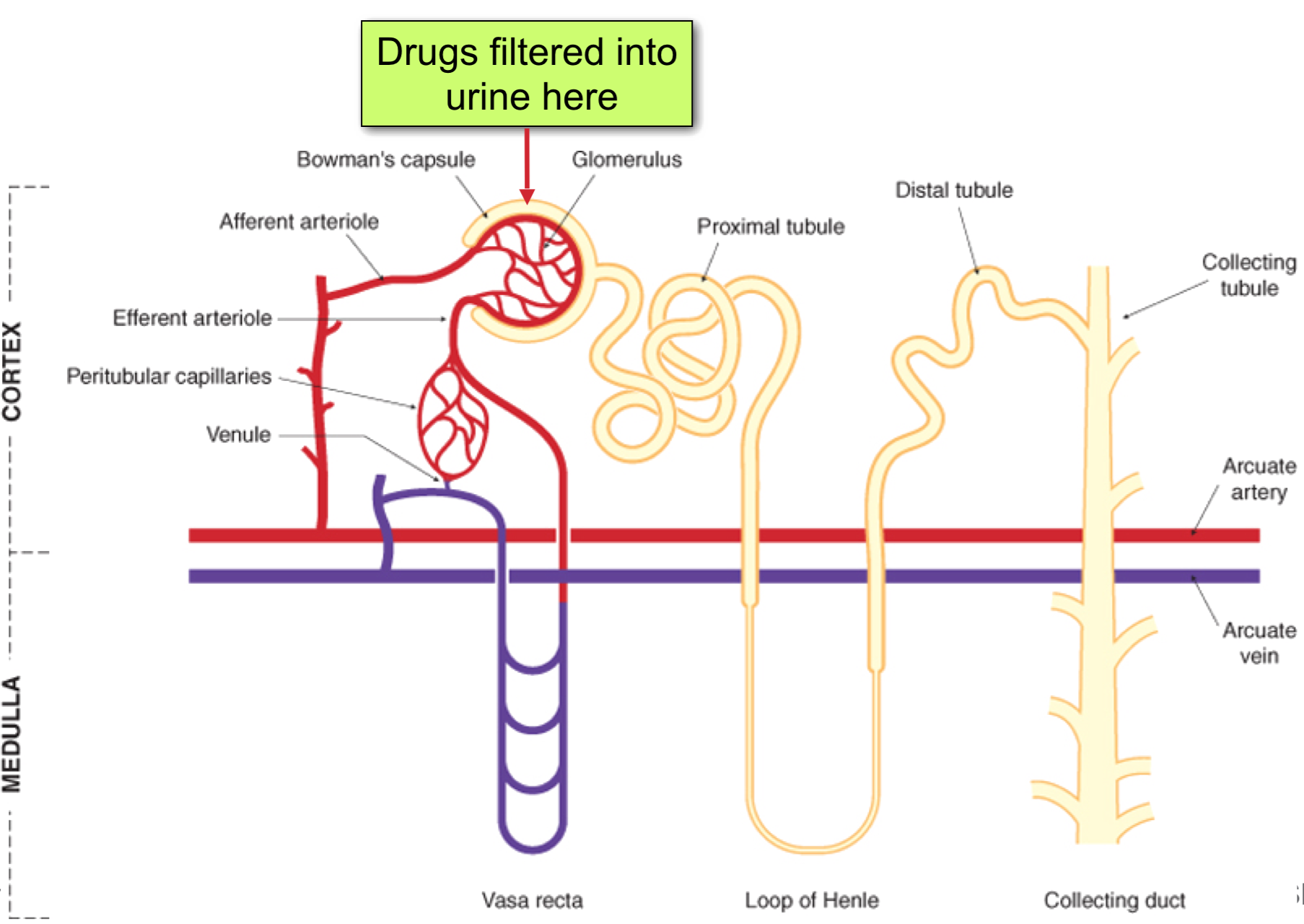
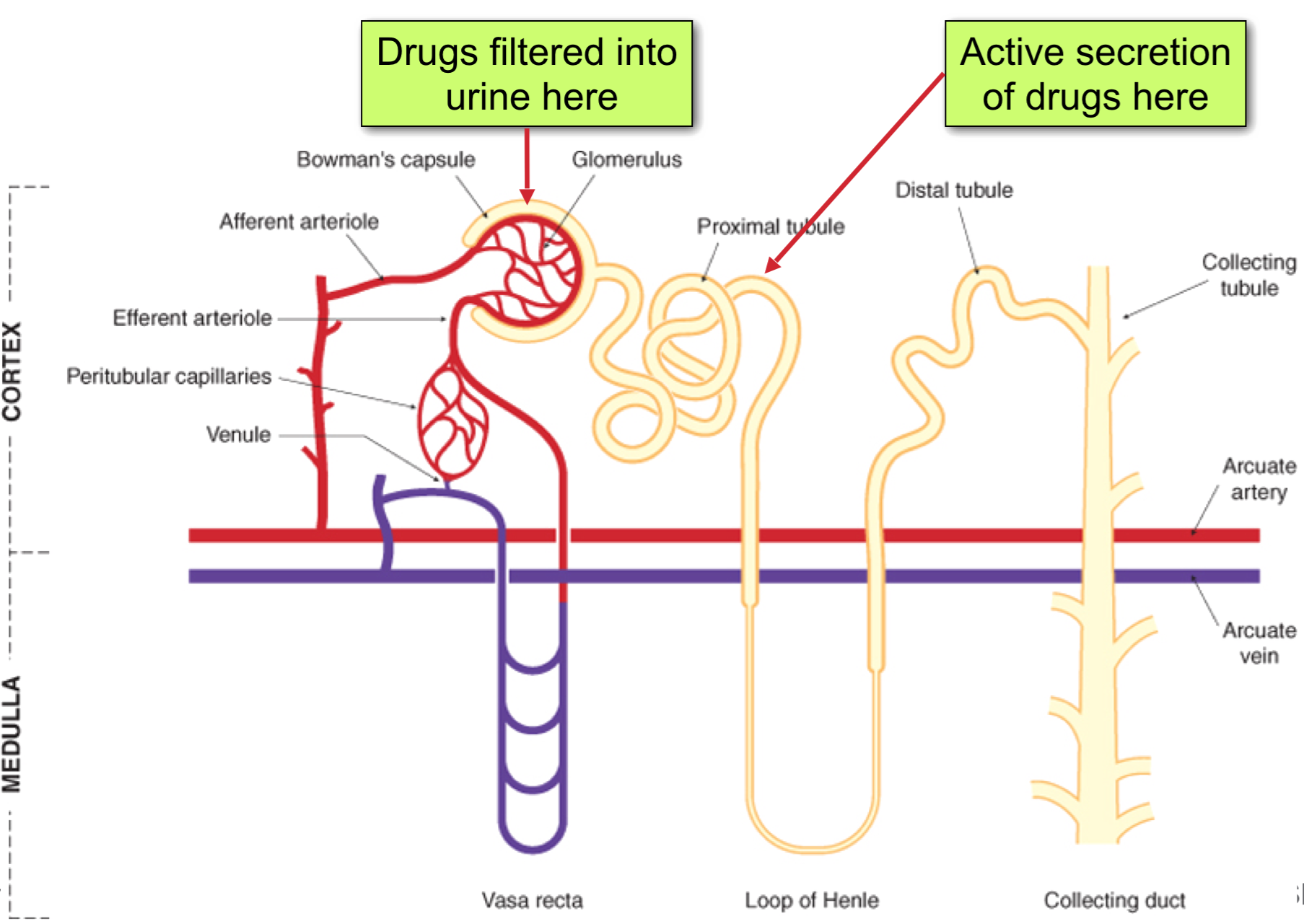
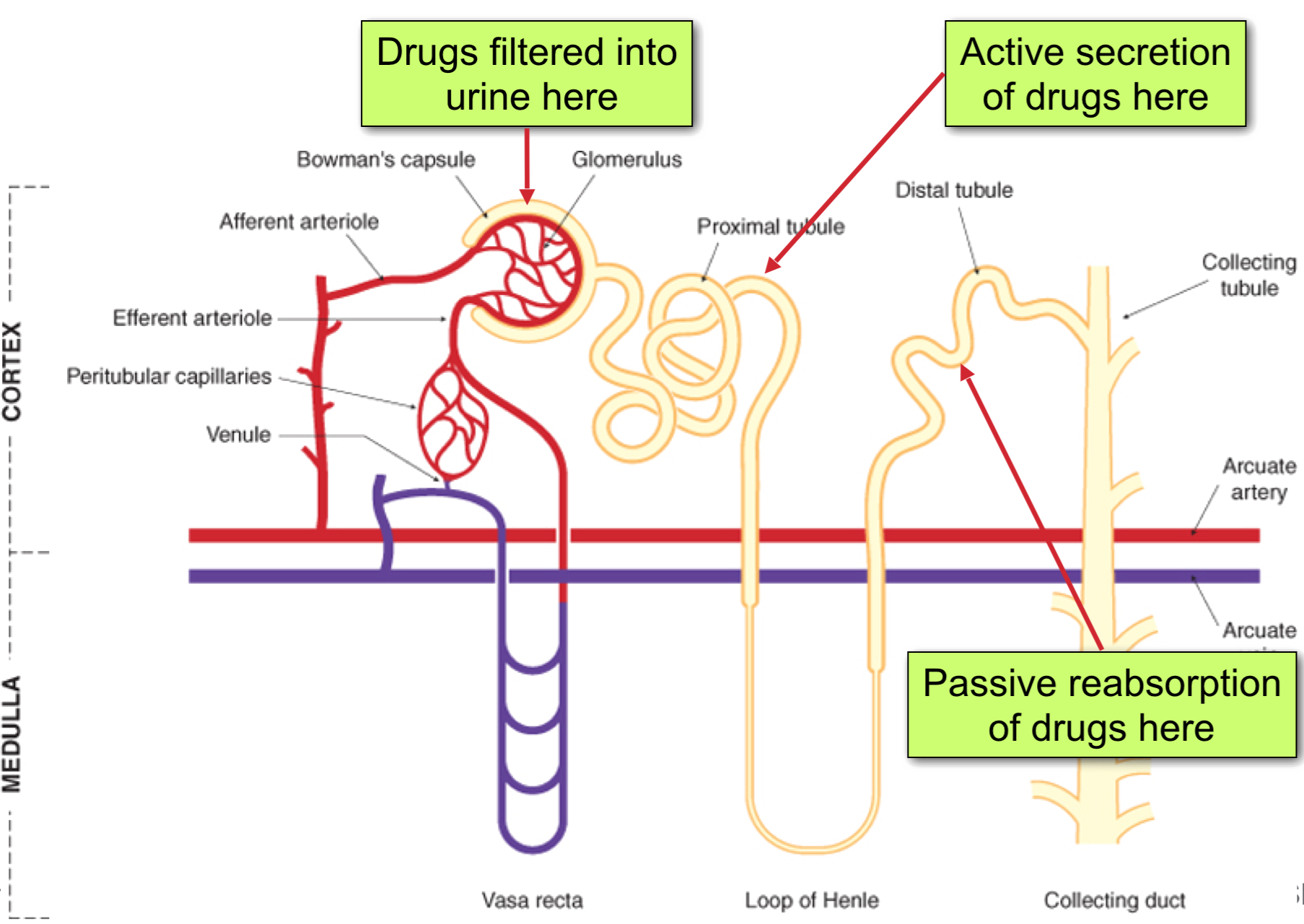
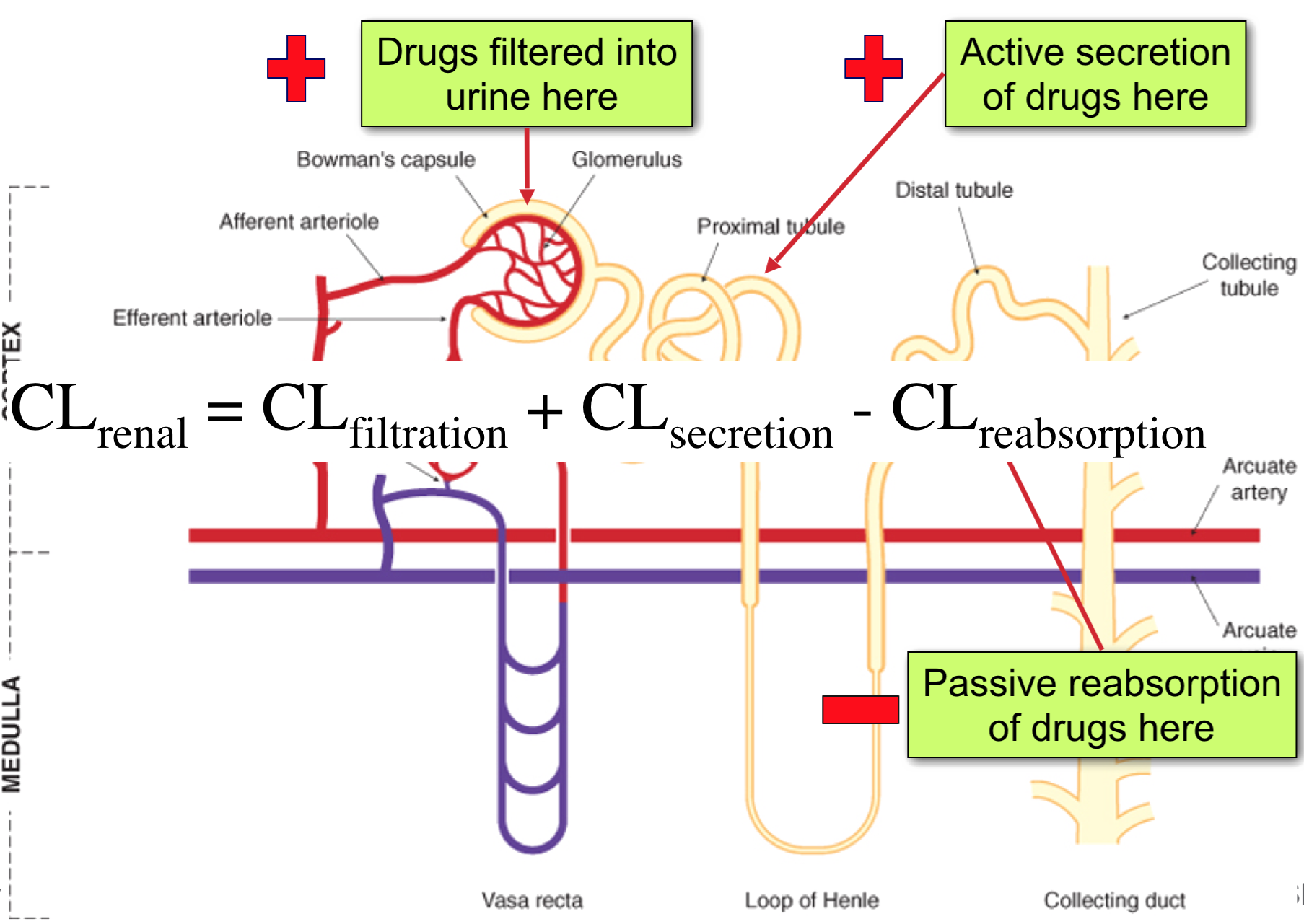
Renal Clearance
CLrenal = CLfiltration + CLsecretion – CLreabsorption
Half life, Volume Distribution, Clearance
Half-life and Vd
Clearance and half-life
Drug excretion 1


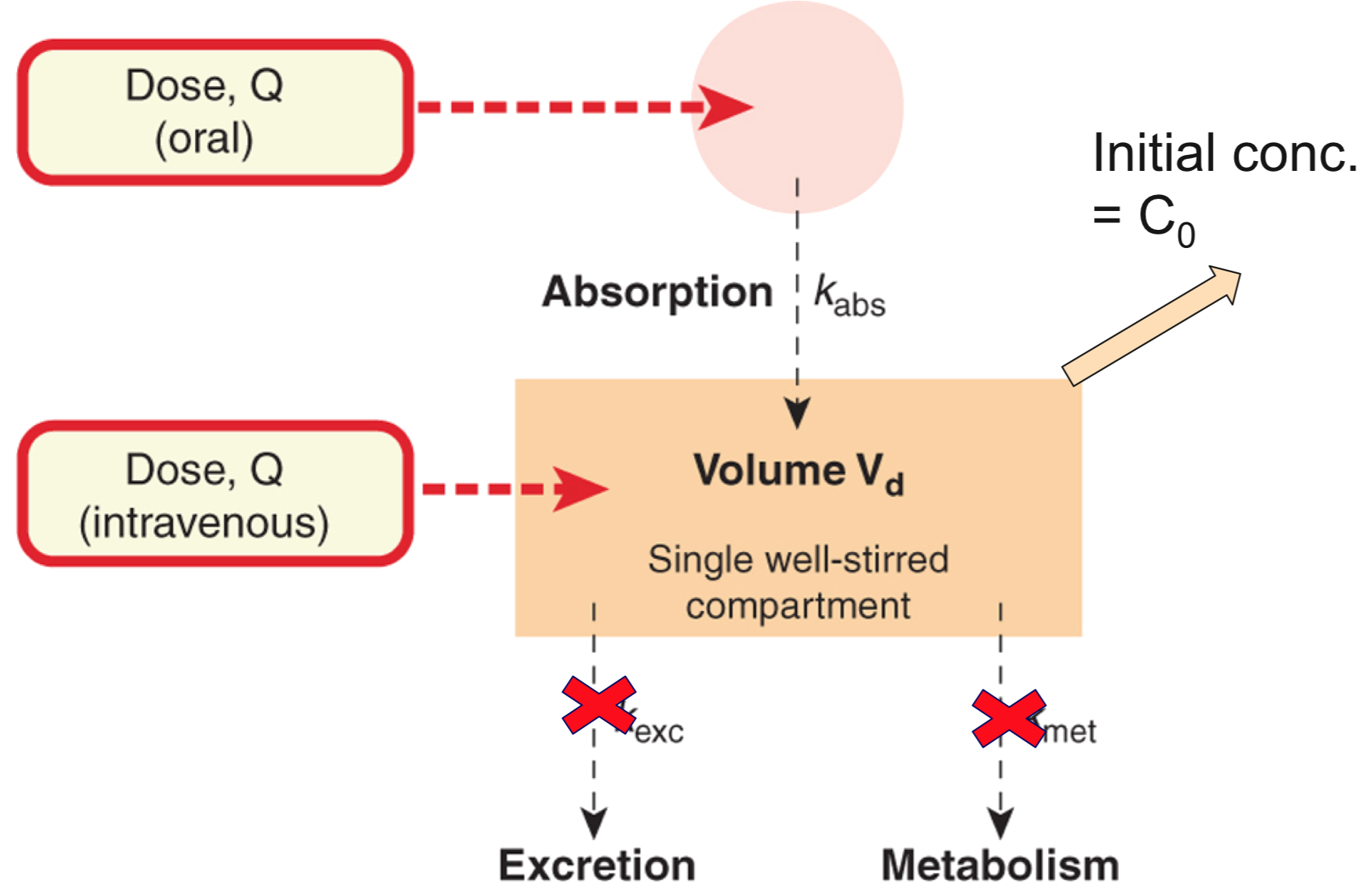
Single compartment model

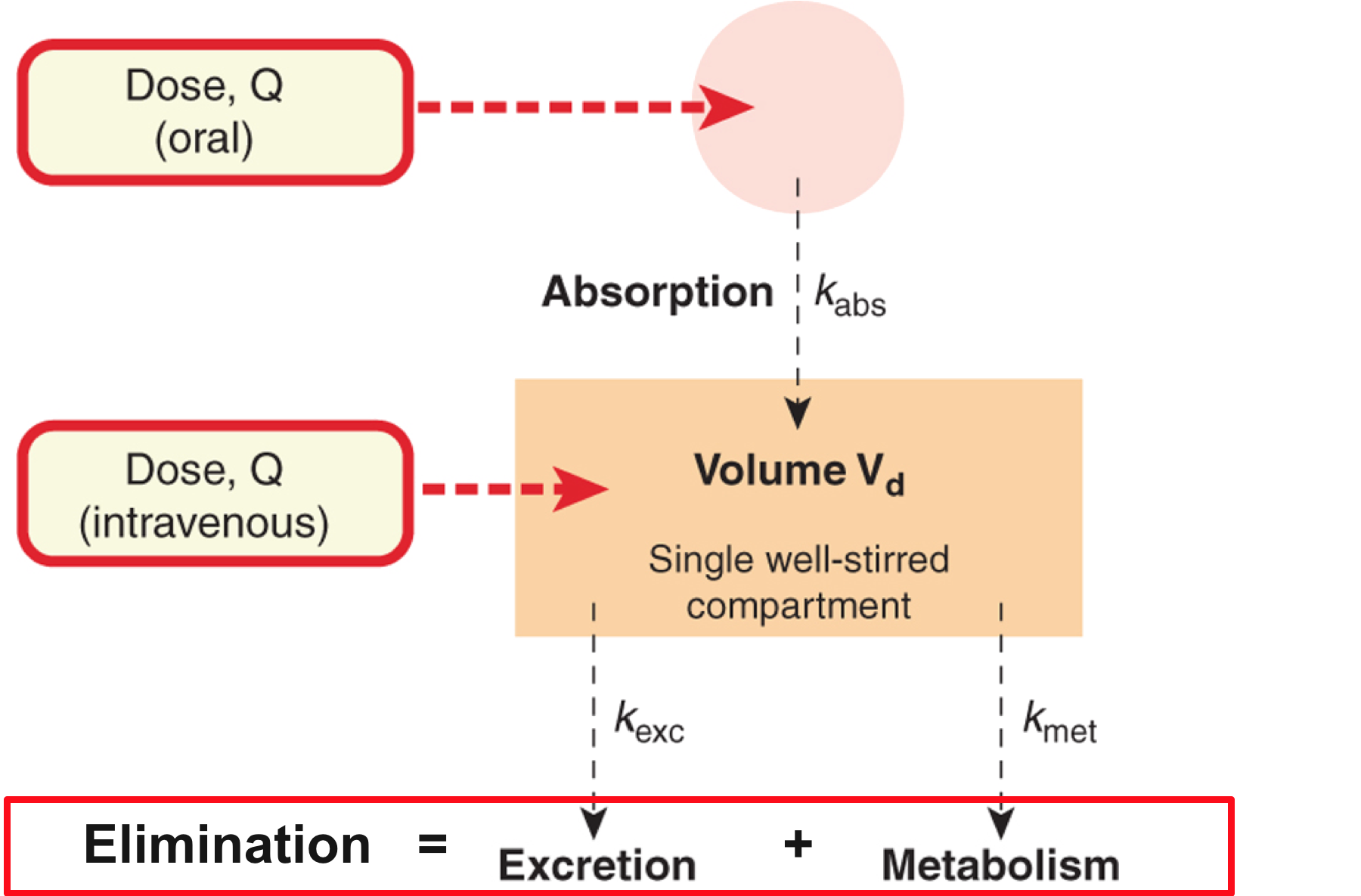
Drug Clearance
Drug clearance 2
- For most drugs, elimination follows an exponential time course with a constant half-life (t1/2) that is independent of plasma concentration (Cp )
- Known as first-order kinetics
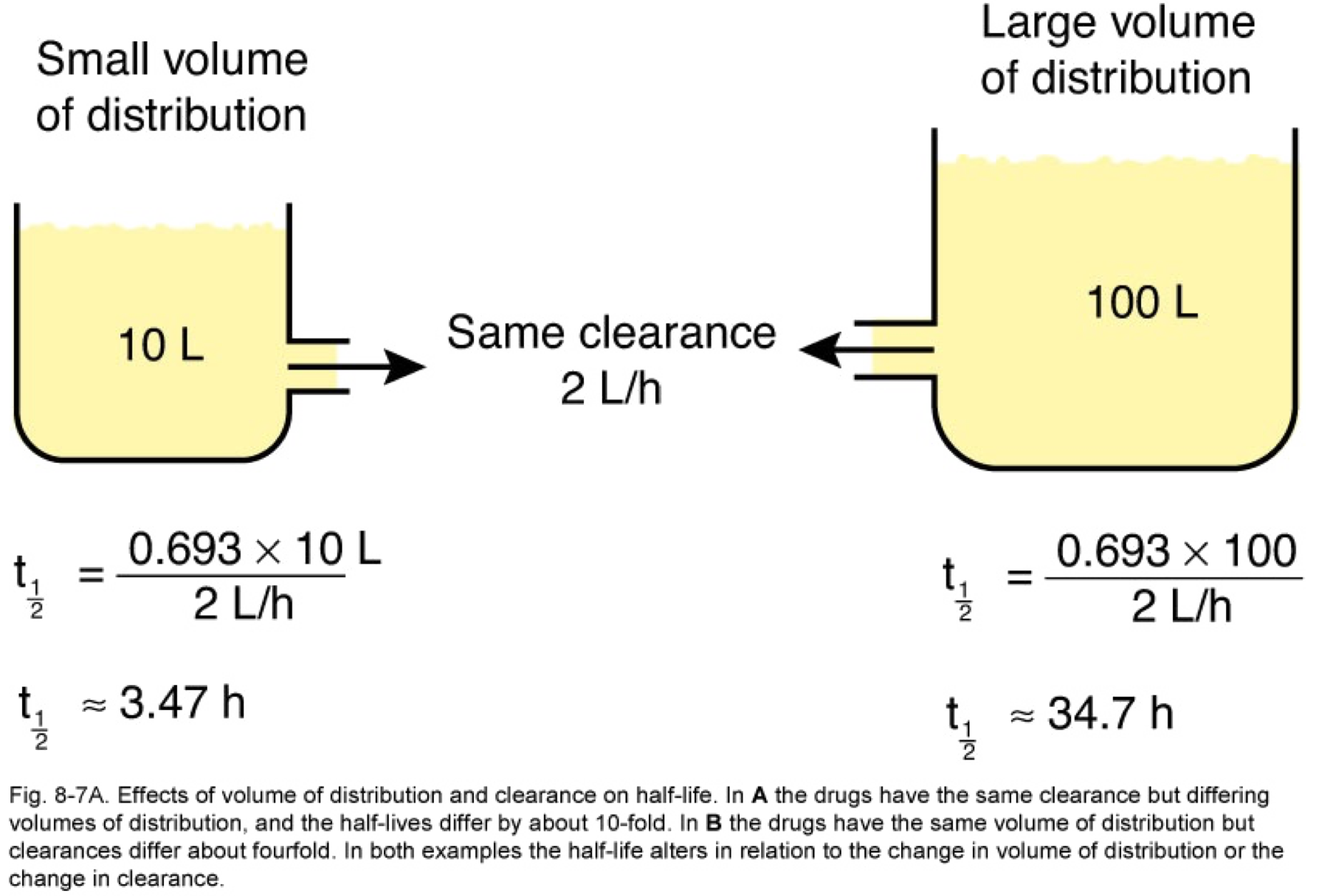
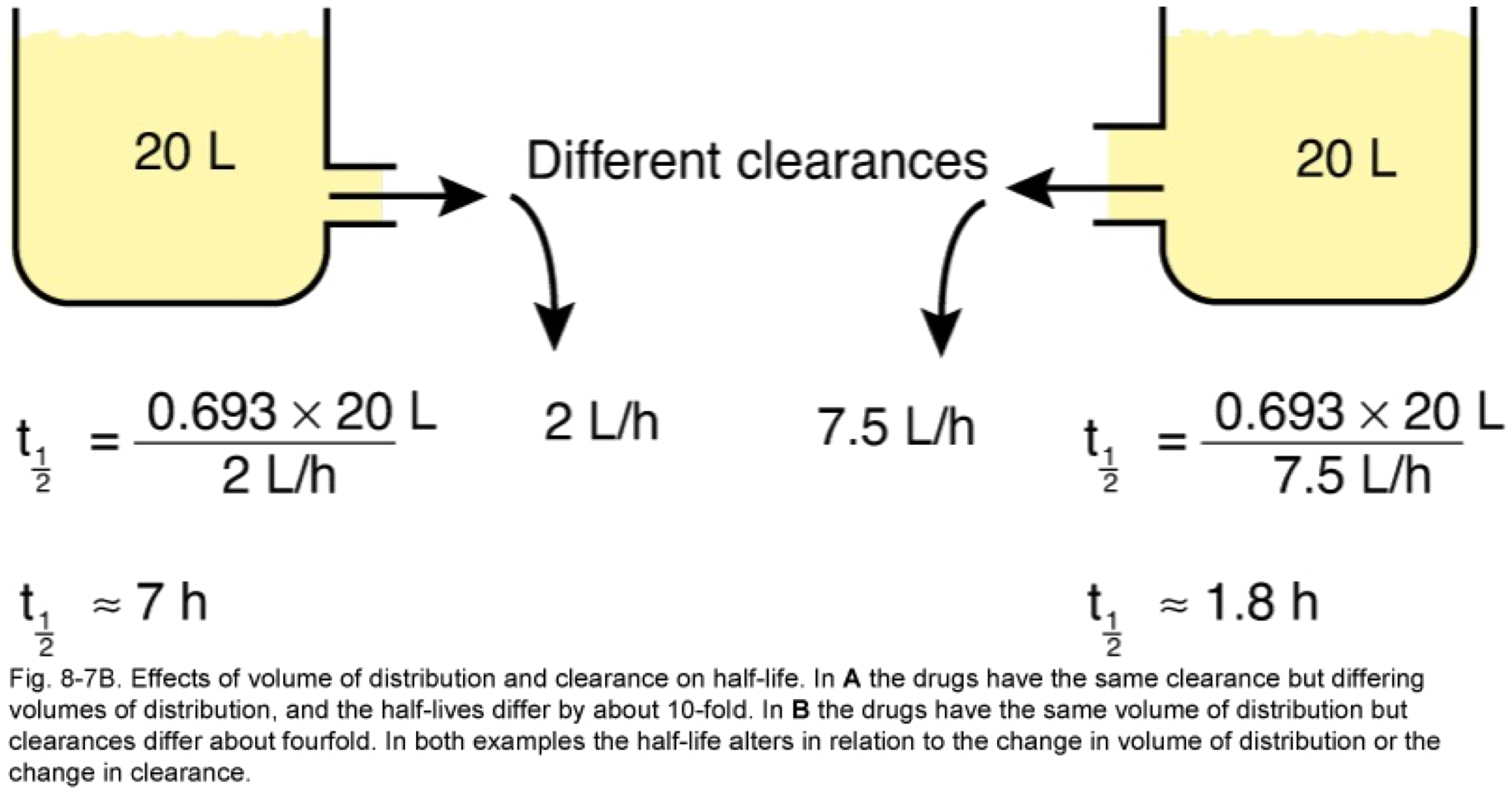
- t1/2 is directly proportional to Vd and inversely proportional to CL
- The initial plasma concentration (C0) = Q/Vd
- Rate of decay described by:
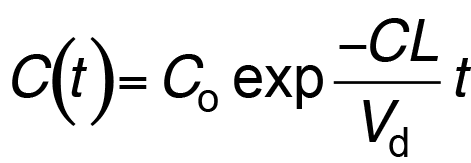
Drug clearance 3
Drug Clearance 3
Clearance is defined as the volume of plasma that is cleared of drug per unit time

Drug clearance 4
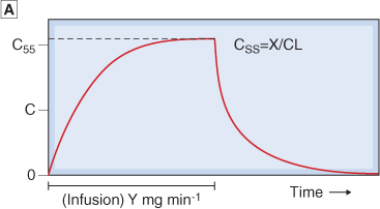
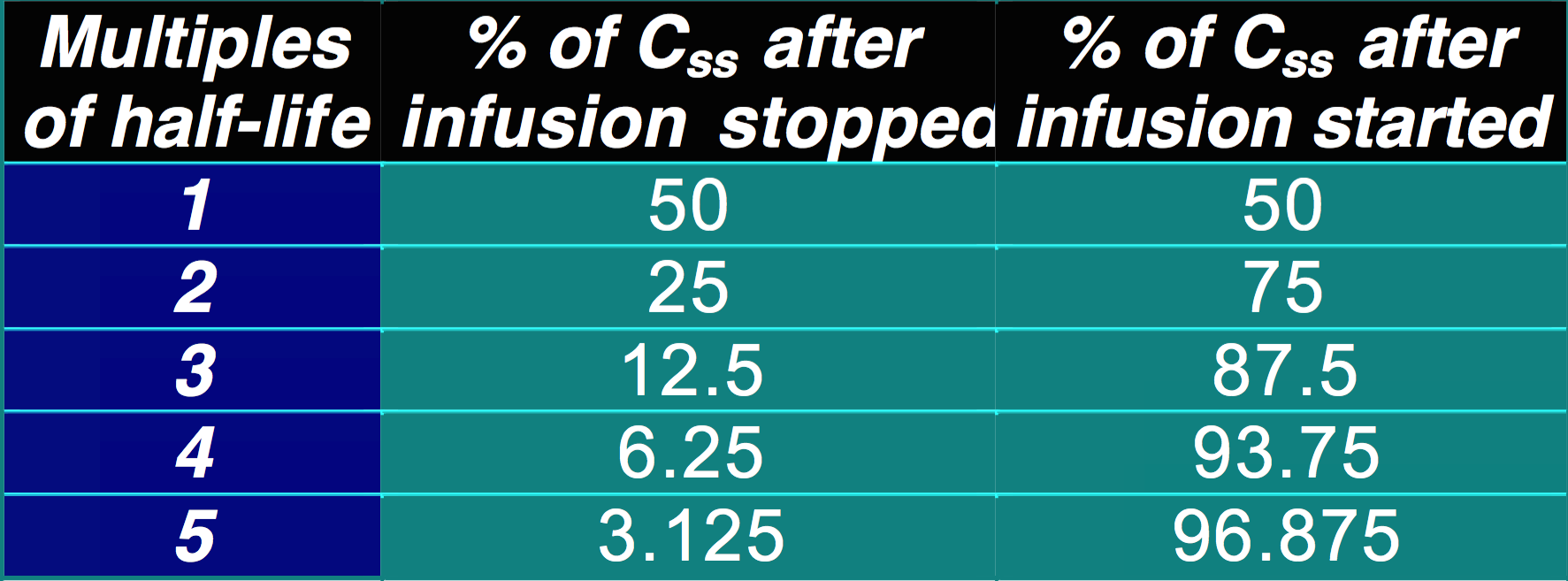
- It takes approximately 4-5 t1/2 for Cp to reach the steady state concentration once an infusion or multiple dosing with the drug is started. The reverse is also true once treatment is stopped:
- ~5% of Css after infusion is stopped (effectively eliminated)
- ~95% of Css after infusion is started
Repeated Dosing
Repeated dosing 1
- Drugs are normally given repeatedly (p.o.) rather than single injection or constant IV
Pharmacokinetics is used to determine:
- What is appropriate dose that must be given repeatedly?
- How often must drug be given to:
- maintain therapeutic levels?
- avoid toxic concentrations?

Repeated dosing – plasma concentrations
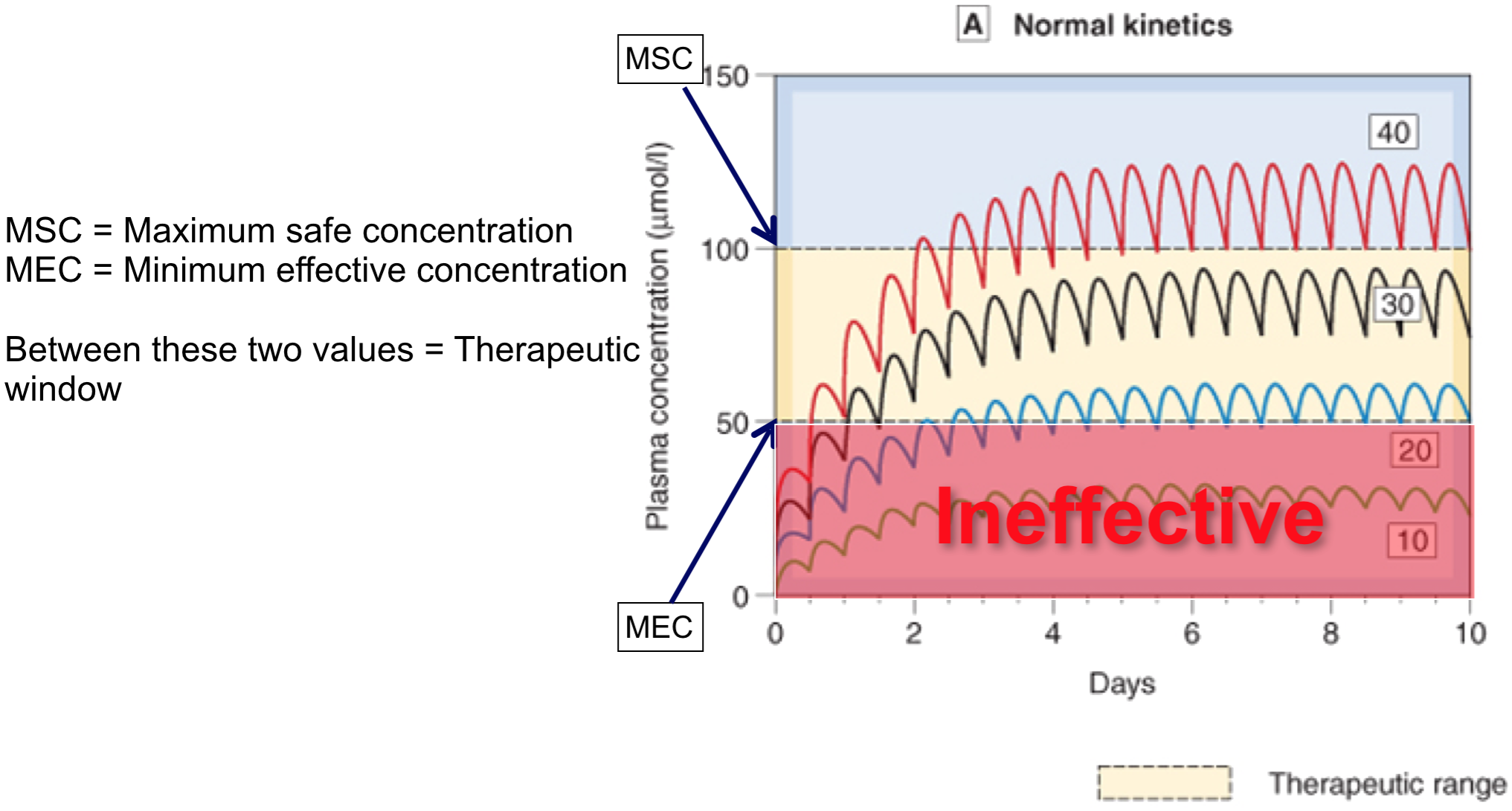
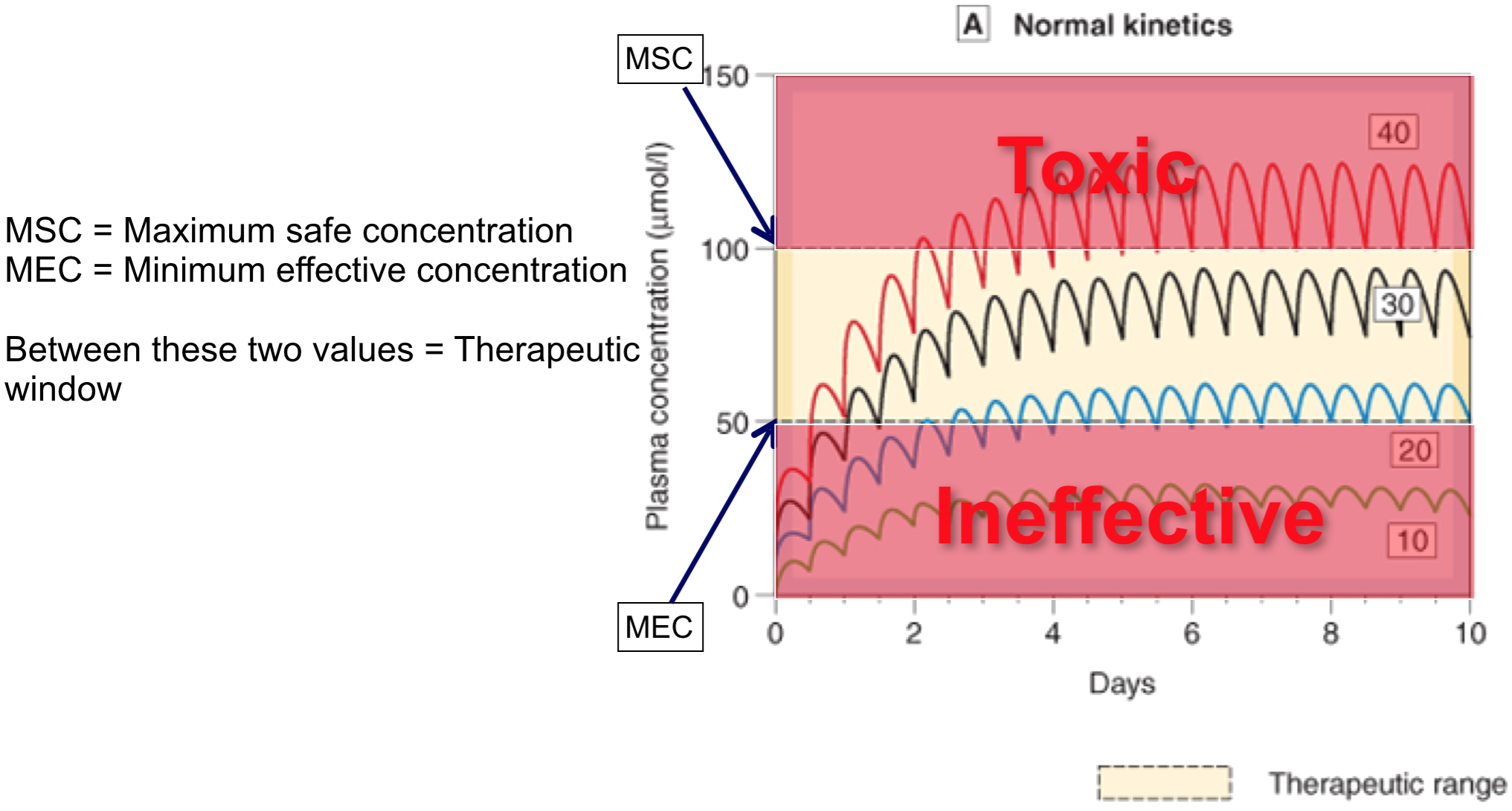
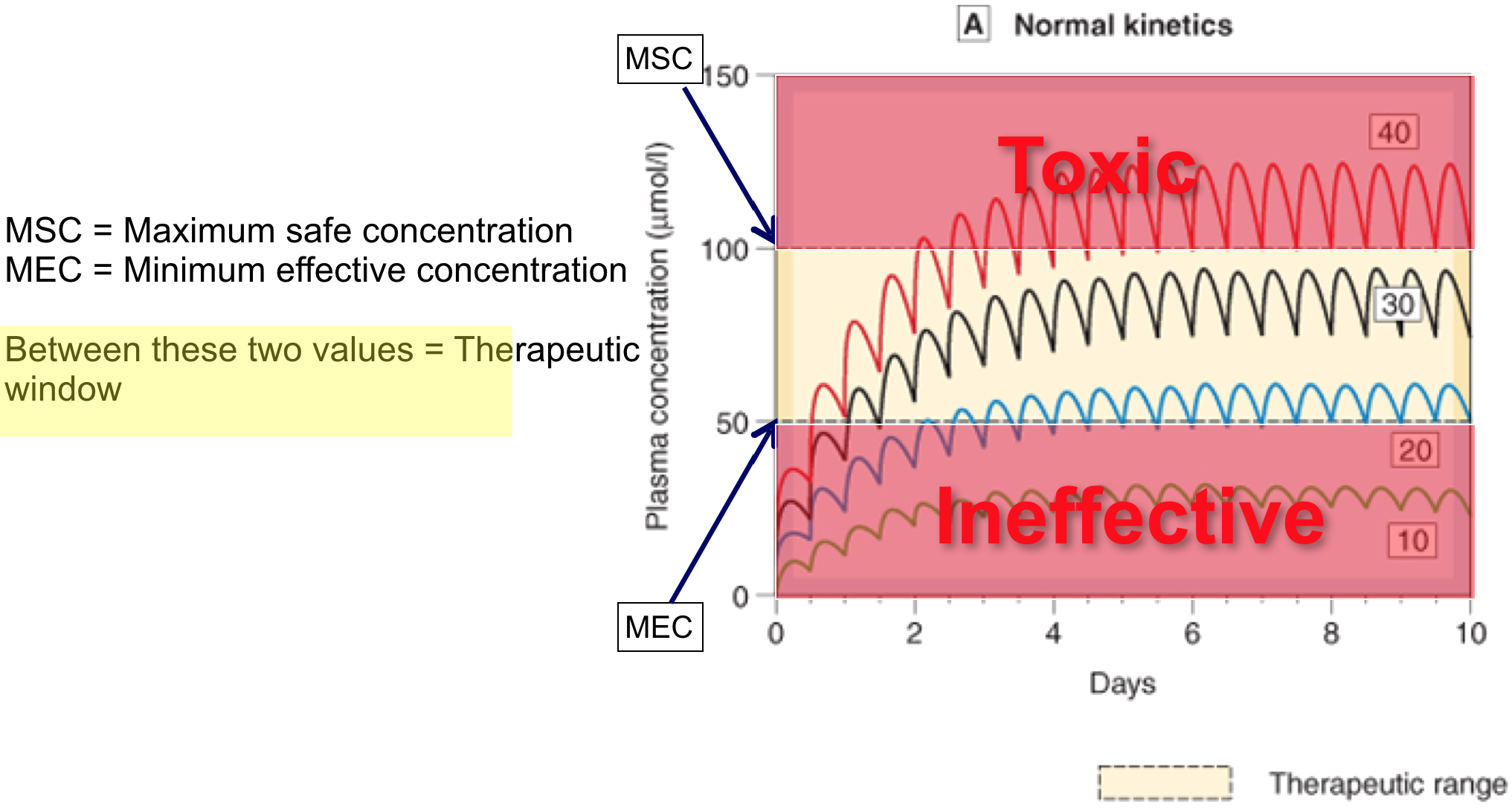
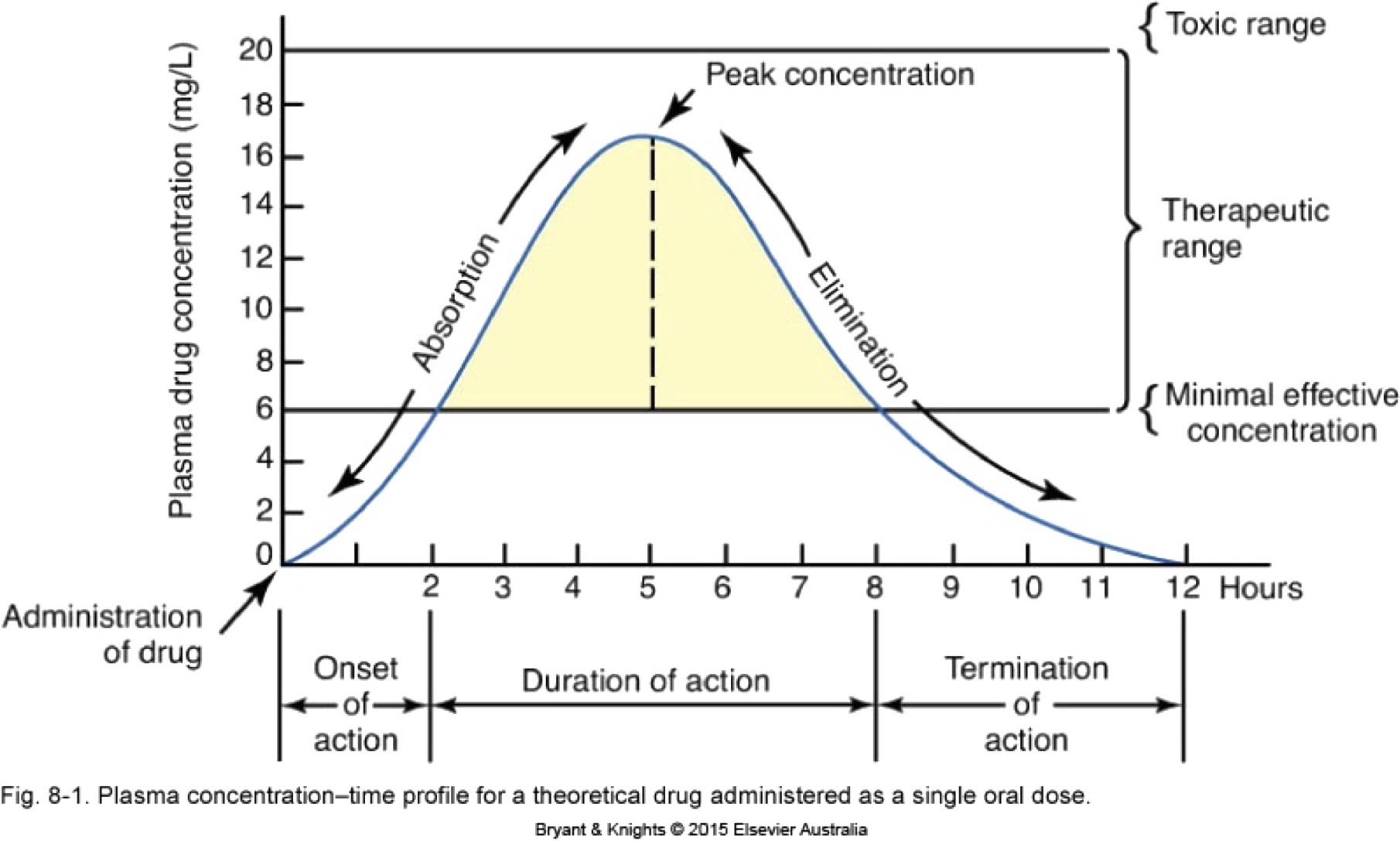
Repeated doses 4
- If dosing interval < 4.5 x t1/2, drug will accumulate (if its greater than this it wont accumulate becase the concentration will come back to zero before the next dose is started)
- With repeated fixed dosing (or IV infusion) it takes 4.5 x t1/2 of a drug to reach max. absorption
- Therefore, after 4.5 x t1/2, rate of drug elimination = rate of drug absorption/supply = maximal accumulation = steady state (Css)
- If you increase the dose of rate of infusion it still takes the same time to reach the steady state it’s just that the steady state plasma concentration will be higher.
Dosing interval 1
- There are two opposing forces at work
- Patient compliance
- Less often a drug can be given, better will be patient compliance – however – the fluctuation in concentration will be greater!

Dosing interval 2
- Fluctuations in plasma concentration
- Fluctuations will oscillate with a range of Q/Vd
- Less often a drug is given, greater will be fluctuations in Cp
- Cp falls below therapeutic levels
- toxic Cp may also be reached

Variations in absorption
- Slower absorption decreases peak Cp and prolongs duration
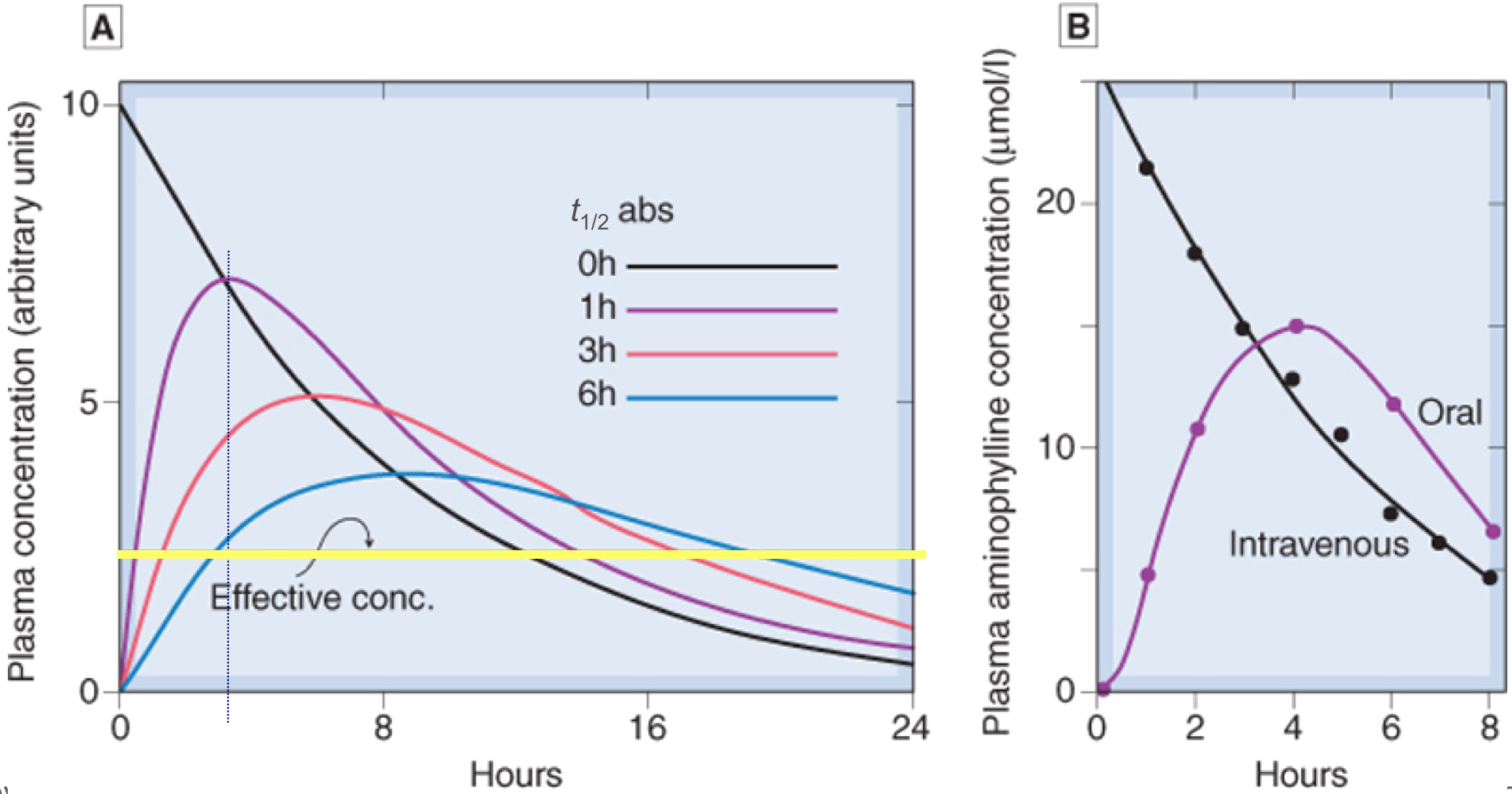
Compartment Models
Single compartment model

Pharmacokinetics 4
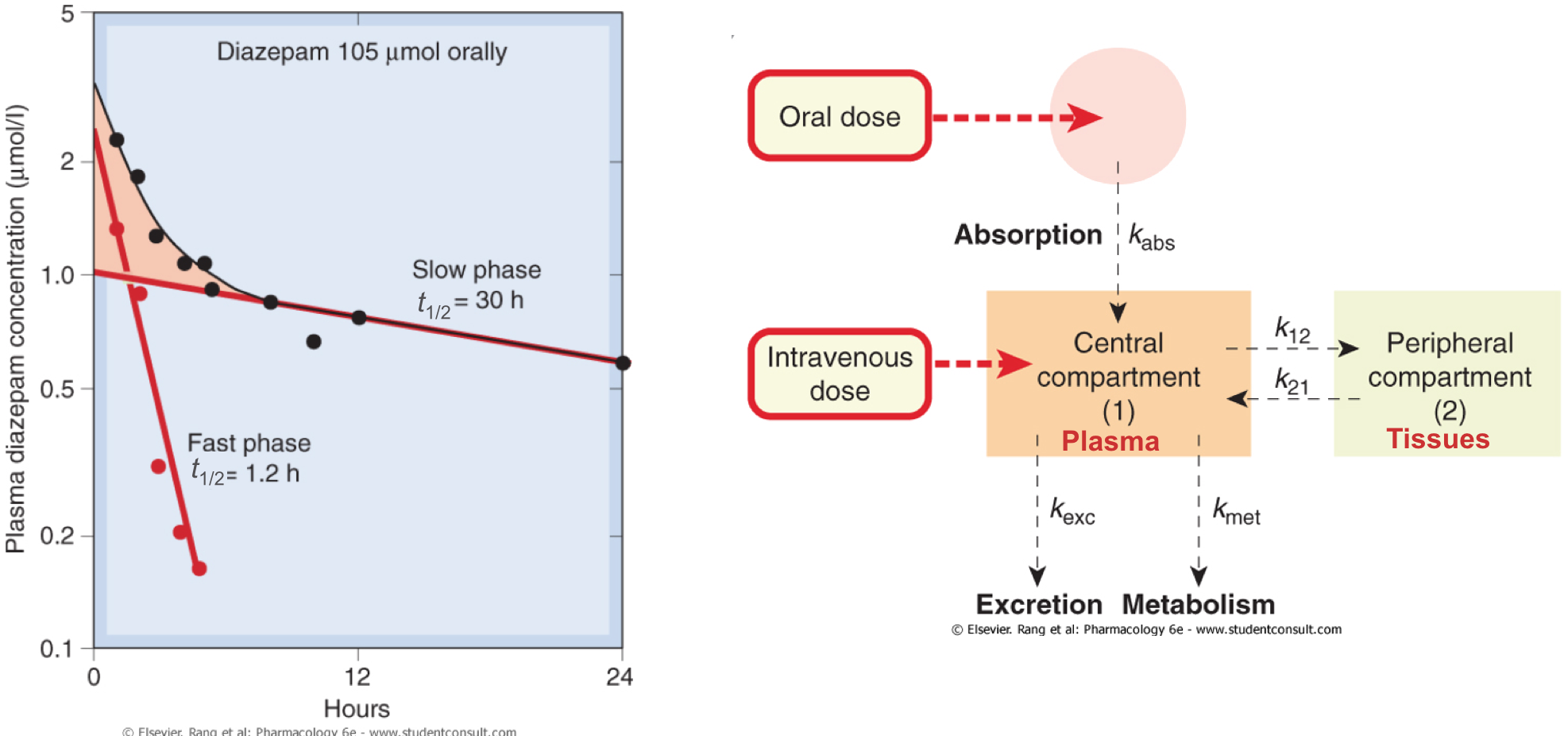
- Very often, however, a fast- and slow- phase of loss of drug from plasma is noted
- The fast phase is characterised as distribution from plasma to the tissues
- The slow phase equates to elimination from the plasma
Two compartment models
- Most drugs are described by a two compartment model
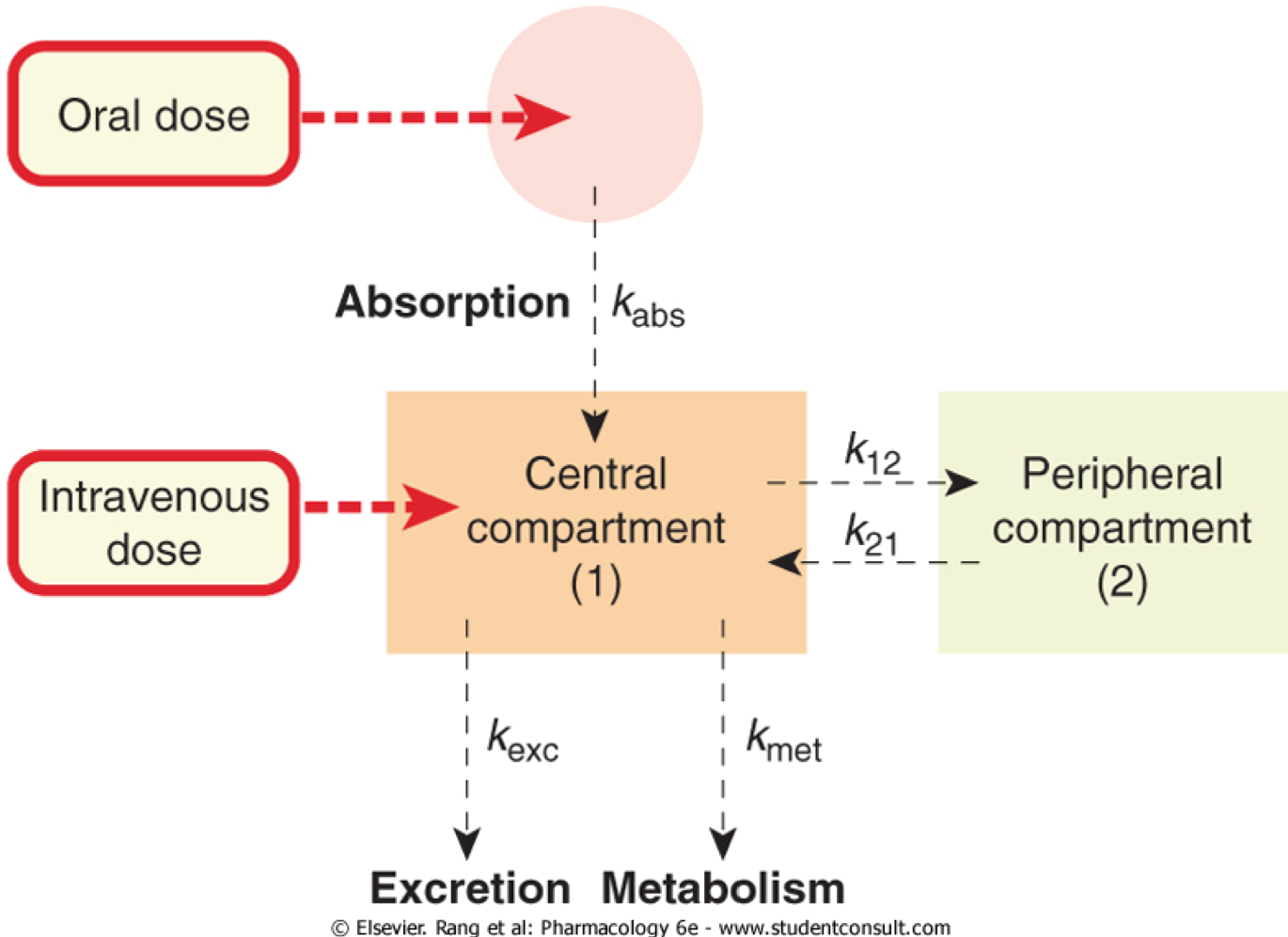
Two compartment models
- Most drugs are described by a two compartment model
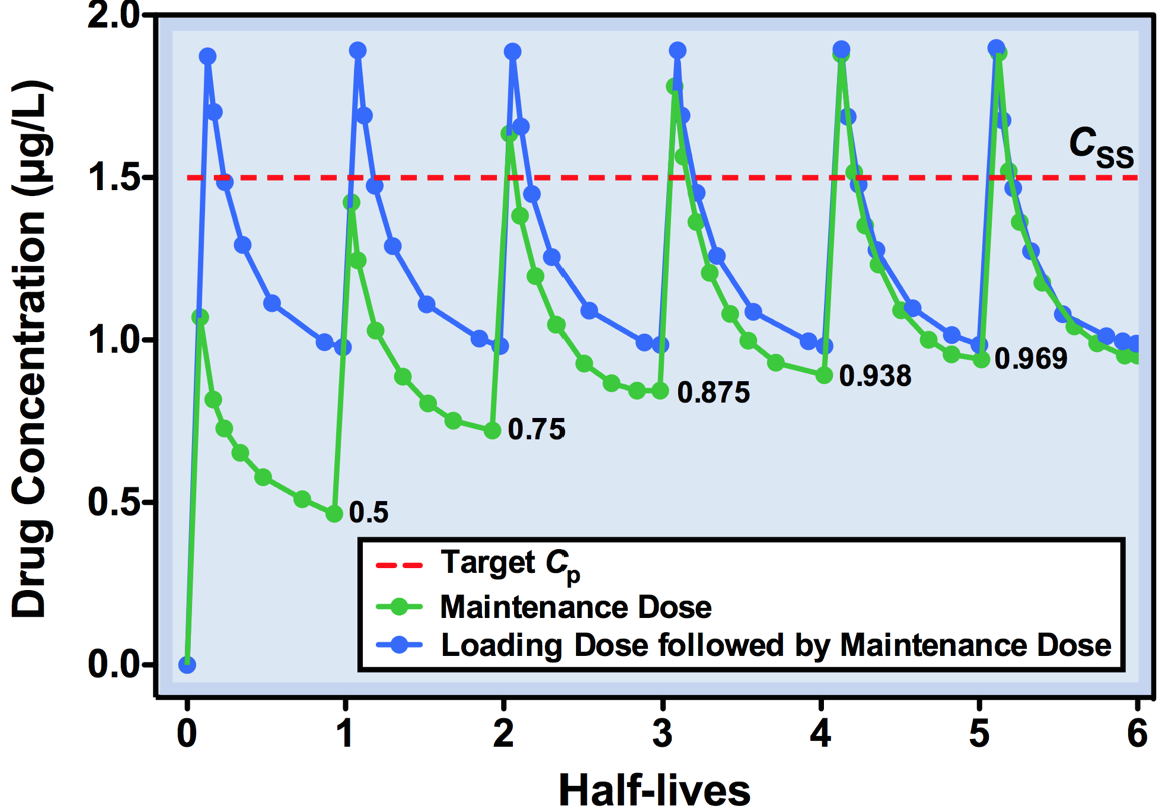
Loading doses
- Sometimes not practical to wait 4-5 t1/2 to attain Css
- Eg. digoxin (t1/2 ≈ 36 hrs), severe CHF needs immediate Rx
- Would take >6 days to achieve Css (less to reach MEC)
- Avoid problem using loading dose (QL) = Vd x target Cp
Dosage Adjustment for Impaired Elimination
Dosage adjustment for impaired elimination 1
- Duration of action and potency of most drugs is dependent on metabolism (hepatic) and/or excretion (renal)
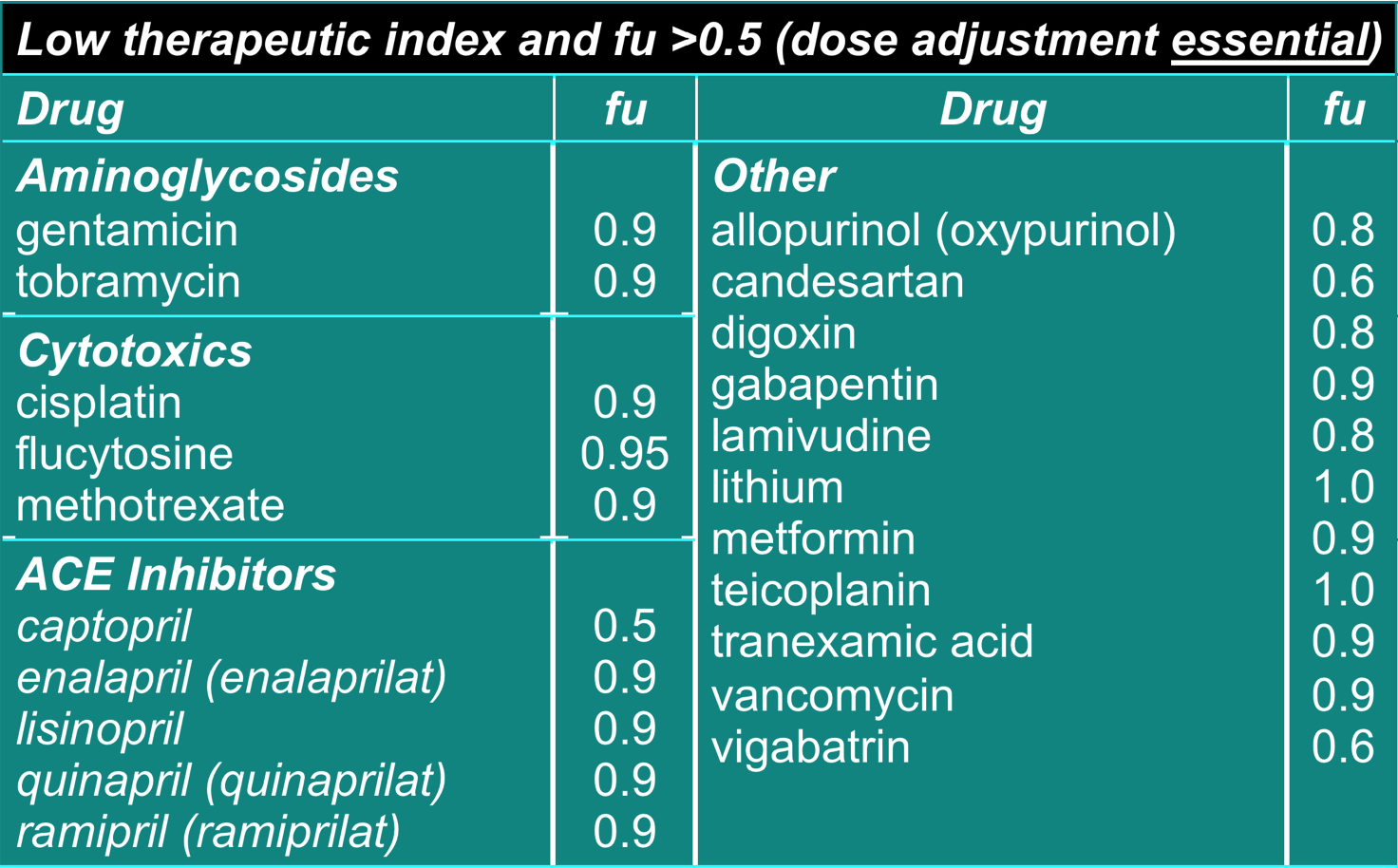
- Ca. 20% of drugs are cleared unchanged through kidneys (eg gentamicin, cisplatin)
- Thus must alter dose of drug given to patients with these dysfunctions
Dosage adjustment for impaired elimination 2

Dosage adjustment for impaired elimination 3
- Dose adjustment required for renal impairment
- ‘fu’ = fraction excreted unchanged
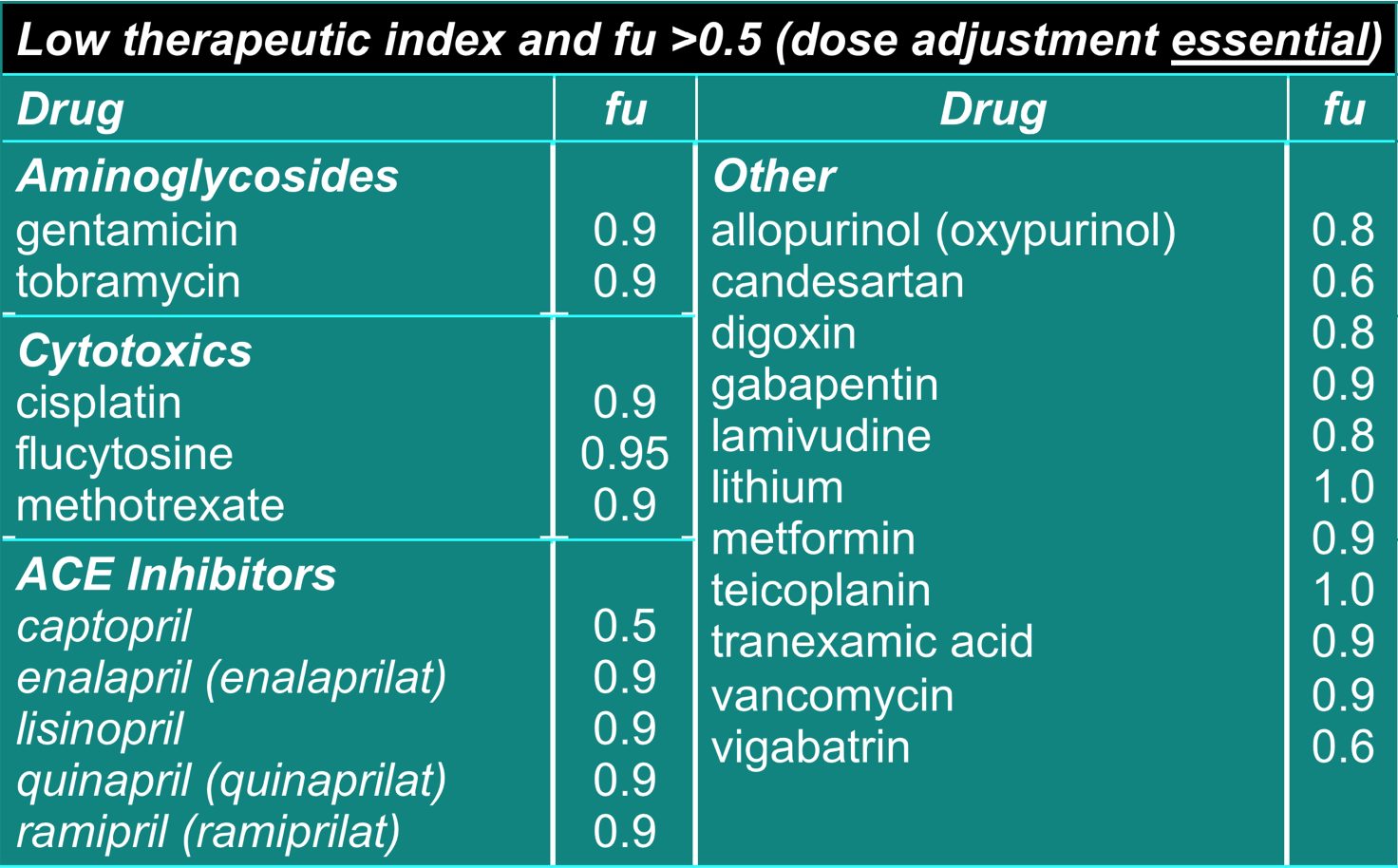
Dosage adjustment for impaired elimination 4
- Dosing in liver disease
- There is no simple measure for hepatic impairment
- Albumin conc and prothrombin index indicate ability to synthesise proteins and thus liver enzymes but only indicate severe, chronic dysfunction
- For albumin <30 g/L (normal 36-55 g/L) and/or raised INR indicate severe liver dysfunction
- High hepatic CL drugs – decrease dose by 50% (see next slide)
- Low hepatic CL drugs – decrease dose by 35%
- ‘Start low, go slow’

Dosage adjustment for impaired elimination 5
- Fraction of drug removed from blood in one passage through liver is the extraction ratio (ER)
- These drugs are subject to first-pass metabolism and altered blood flow (eg in CHF)
Zero-order Elimination
Zero-order elimination 1
- Theoretically all drugs could saturate their metabolising pathways (eg. enzymes or pumps)
- For most drugs this occurs above therapeutic concs (eg only with overdose)
- For a few drugs this occurs at therapeutic concs
- This is known as zero-order elimination (saturable kinetics)
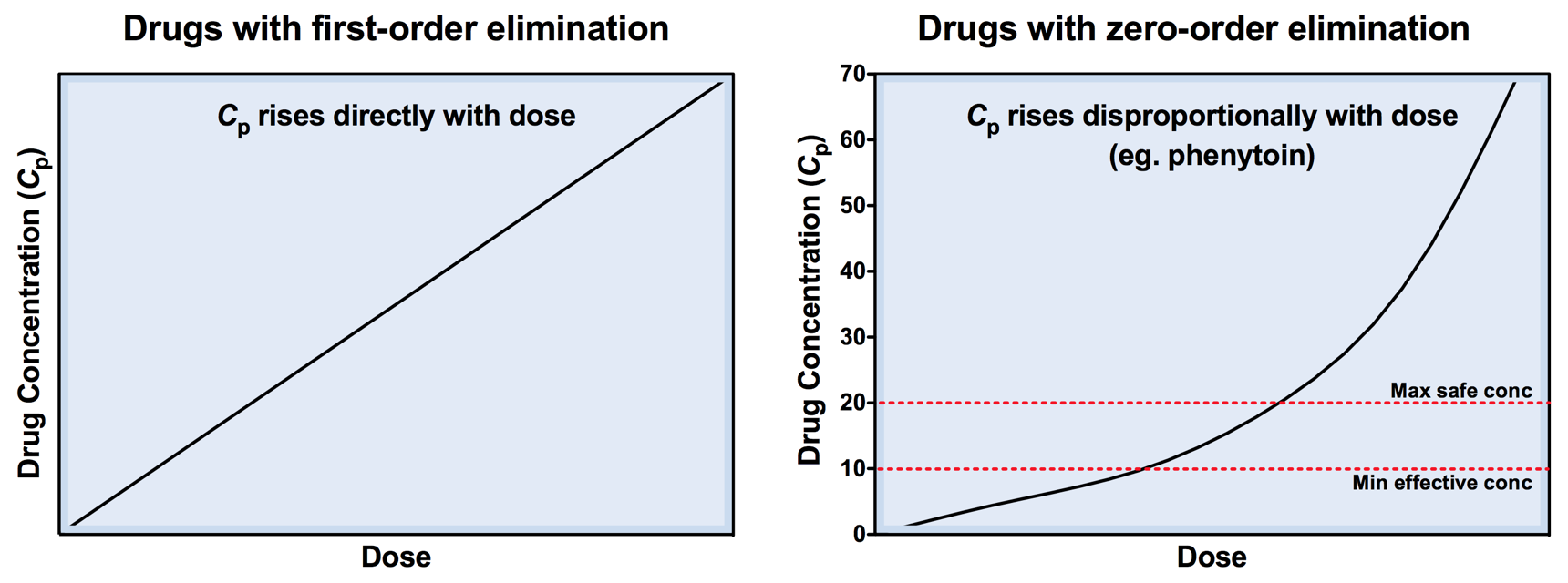
Zero-order elimination 2
- Below Km, rate of elimination is proportional to Cp
- Above Km, rate of elimination becomes independent of Cp
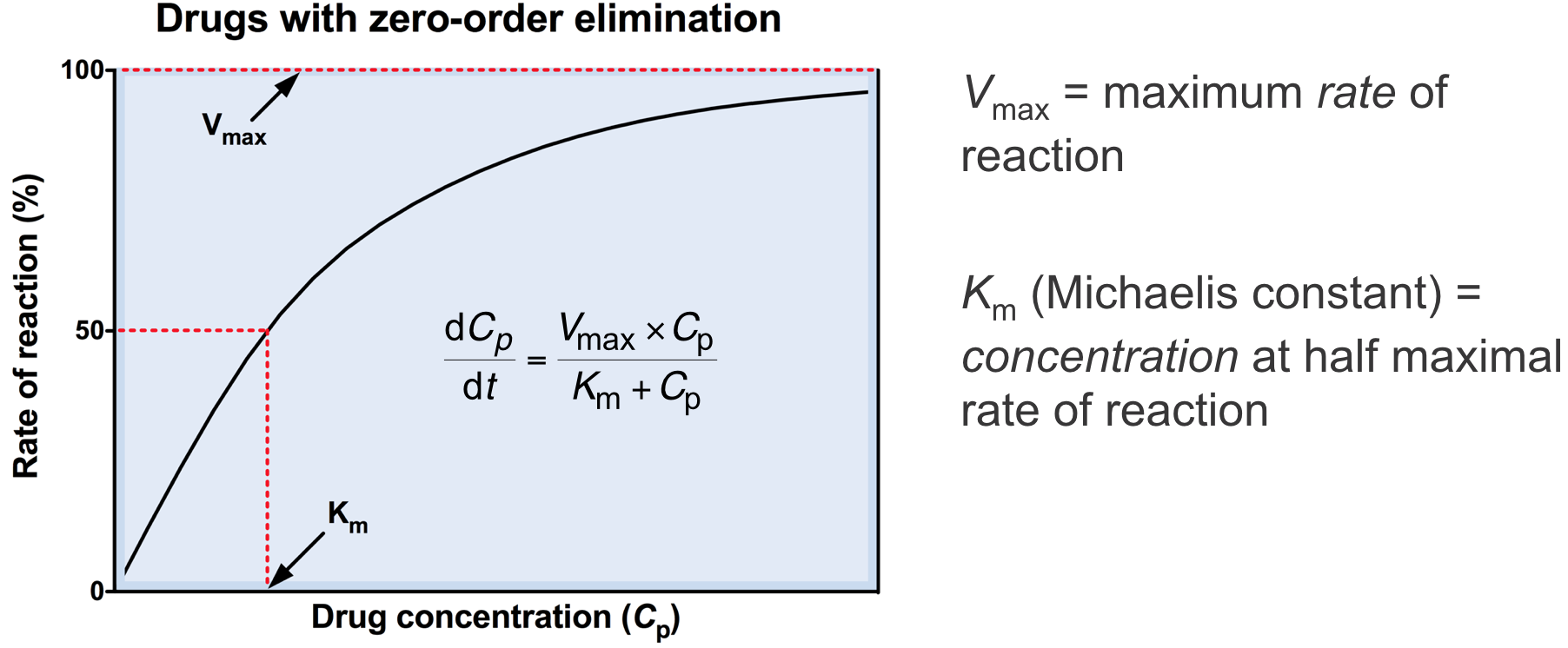
Zero-order elimination 3
- Examples of drugs undergoing zero-order elimination include:
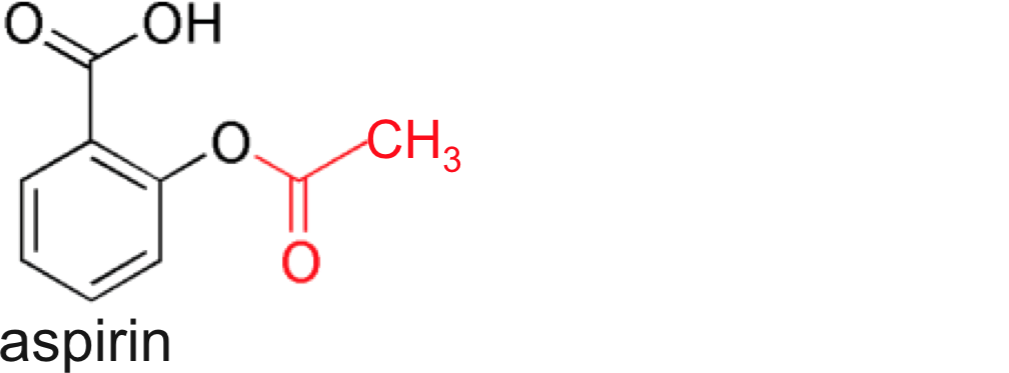
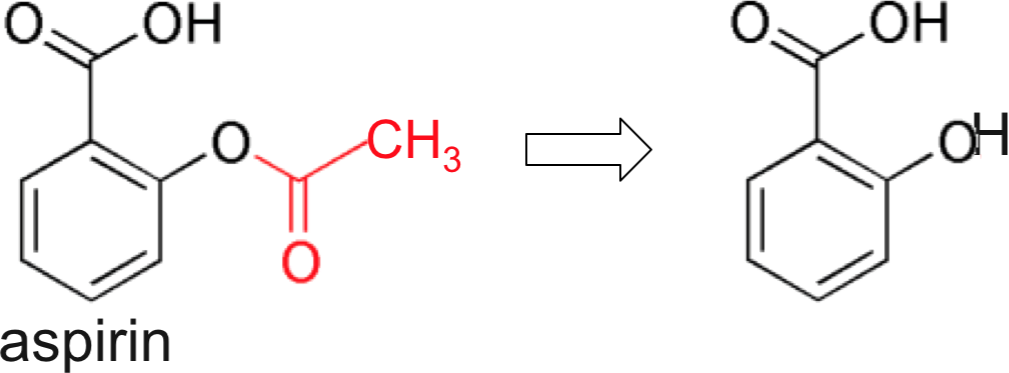
- Aspirin (saturates with one to two 300 mg tablets)
- Coumarins
- Ethanol (Km = 0.01%, legal limit for driving = 0.05%)
- Quinidine (narrow therapeutic index)
- Heparin
- Tetracycline
- Amylobarbitone
- Phenytoin (narrow therapeutic index, Km 7 mg/l, therapeutic range 10-20 mg/l)
- Can also occur in overdose situations, with high Cp
Zero-order elimination 4
- Rate of eliminated is independent of drug starting concentration
- t1/2 is dependent on drug starting concentration
- Eg. Ethanol = 4 mmol/hr
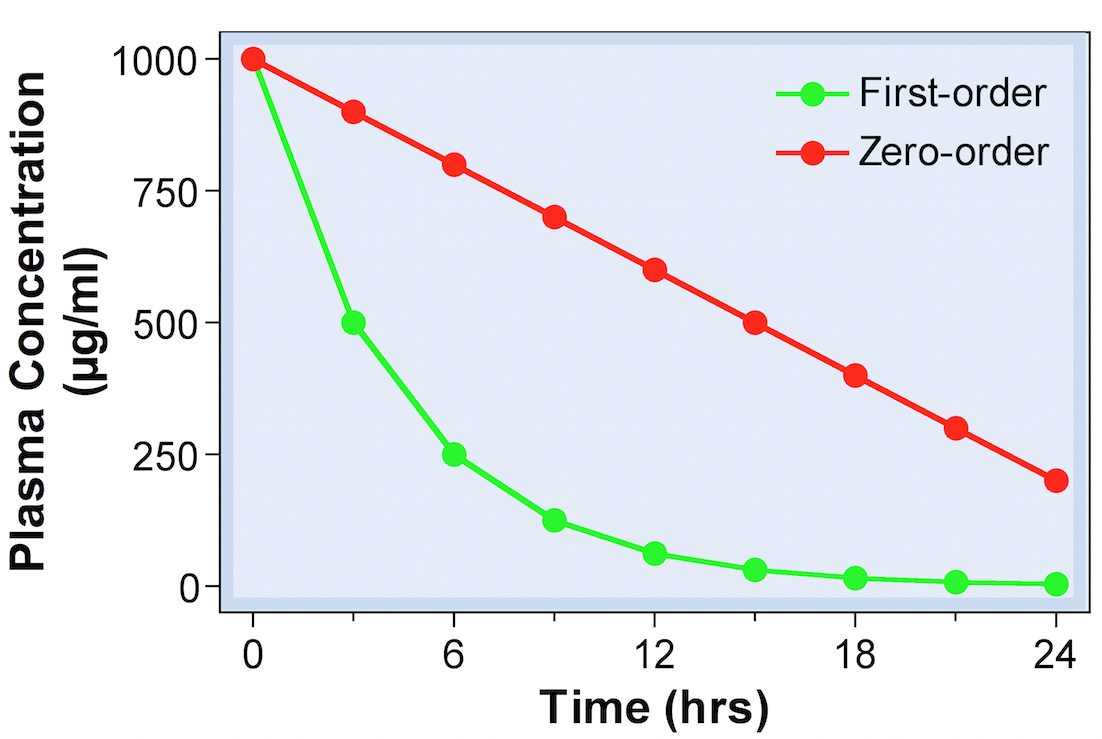
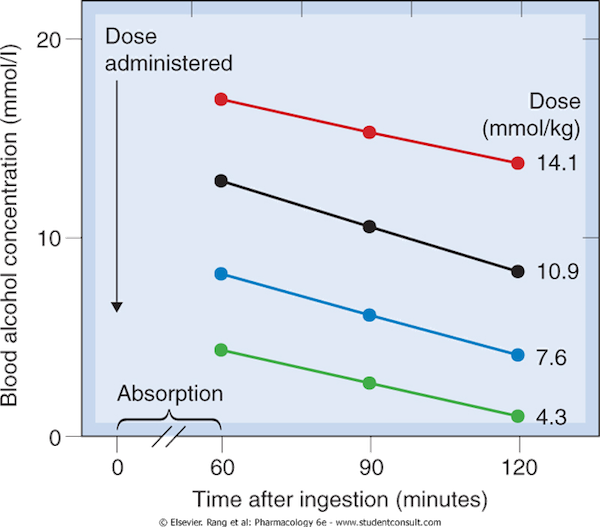
Zero-order elimination 5
- Once the Cp producing the maximal rate of metabolism is exceeded, the Cp will, in principle, increase indefinitely and a steady state Cp will not be reached
- Therefore, Cp should be regularly measured
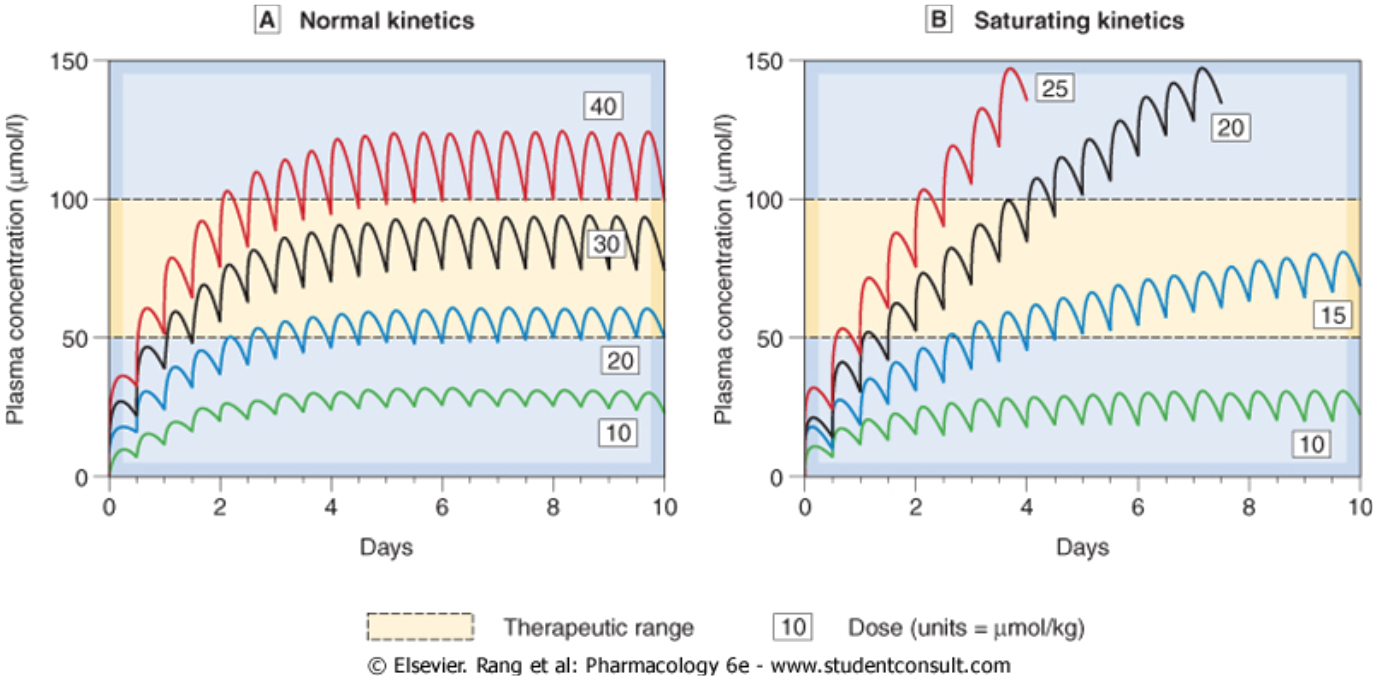
Questions
Question
Drug Y has a steady-state plasma concentration of 1000 mg/L, a half-life (t1/2) of 1 hour and undergoes first-order elimination. What will be the approximate plasma concentration (Cp) 4 hours after i.v. infusion is discontinued?
(A) 10 mg/L
(B) 250 mg/L
(C) 40 mg/L
(D) 62.5 mg/L
(E) cannot be answered without knowledge of the dose given