A/Prof. Ken Rodgers School of Life Sciences
Learning Objectives
- Understand the concept of prodrugs
- Outline how the metabolism and/or excretion of a drug may be influenced by the physicochemical properties of the drug
- Describe the basic mechanisms of Phase 1 and Phase 2 reactions
- Outline the mechanisms involved in renal and biliary elimination of drugs
- Describe how enterohepatic recirculation prolongs the duration of action of drugs
References
- Rang HP, Dale MM, Ritter JM, Flower R and Henderson G (2015) Pharmacology, 8th Edition, Churchill Livingstone, Sydney.
- Drug metabolism and elimination – Chapter 9
Drug Elimination
- Irreversible loss of drug from the body is a combination of 2 processes
- Metabolism
- Excretion



Drug Metabolism: Outline
Drug metabolism: definition
- Metabolism – Enzymic conversion of one chemical entity to another in the body
- Before excretion through the urine, most drugs undergo metabolism in the liver


Drug Metabolism
- Terminates drug action (treated as a xenobiotic)
- Allows for more rapid elimination of drug
- Most occurs in the liver via microsomal (smooth endoplasmic reticulum) and non-microsomal (mitochondria, soluble) enzymatic reactions
- Lipophilic drugs have polar / charged groups added in liver
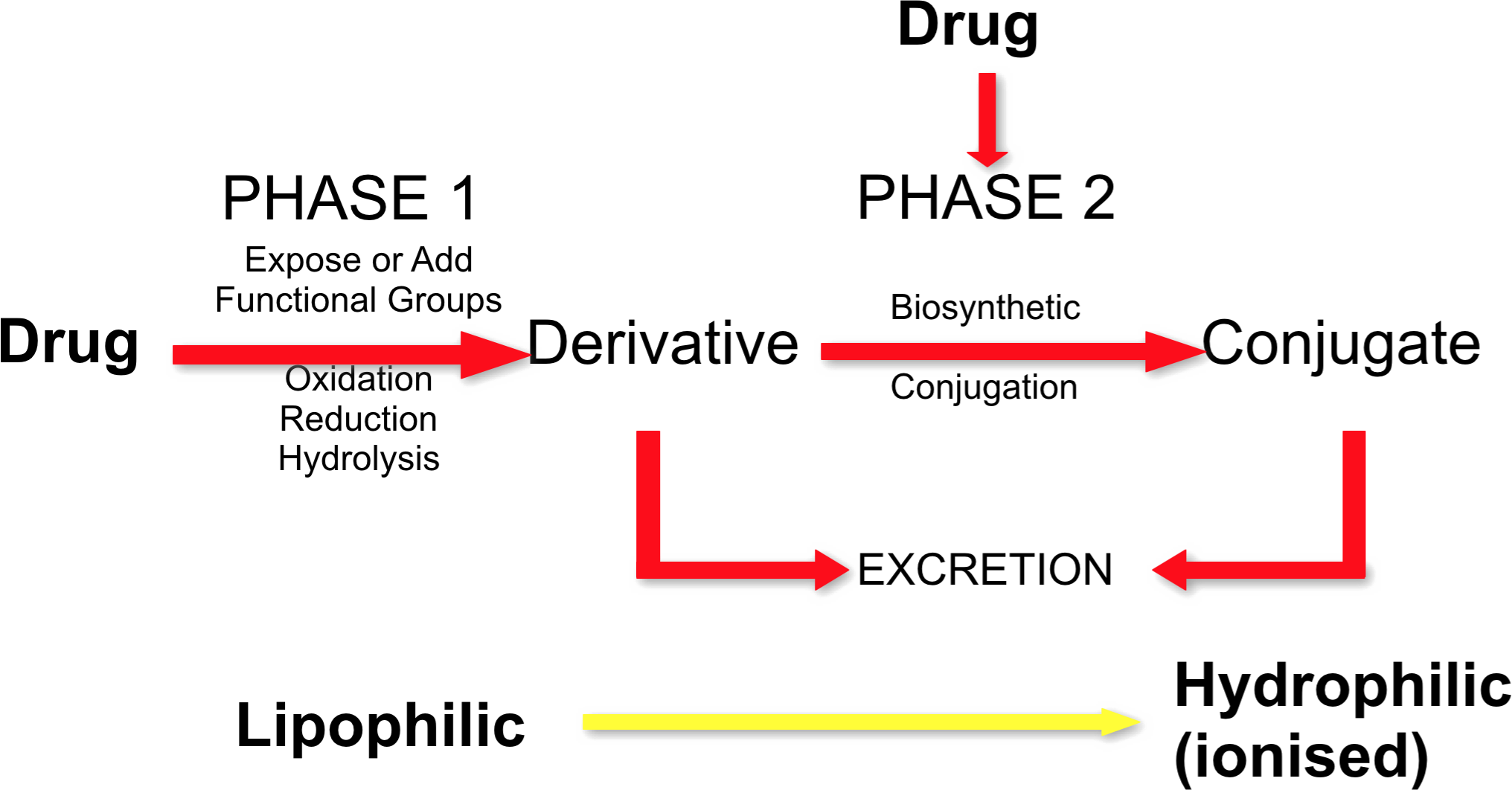
Phases of Drug Metabolism
- Metabolism involves two types of biochemical reaction
- Phase 1 reactions (predominate)
- Oxidation, reduction, or hydrolysis
- Catabolic reactions (breakdown)
- Generate or expose a functional/reactive group. Products can be more reactive or toxic than precursor
- Phase 2 reactions
- Conjugation with hydrophilic groups
- Anabolic reactions (build up)
- Usually results in inactive compounds
- Phase 1 reactions (predominate)
Drug Metabolism: Phase 1
Drug Metabolism: Phase 1
- Phase 1 reactions
- Often involve mixed function oxidase system
- Cytochrome P-450 plays most important role
- Often introduce a reactive group to the molecule
- Adds or exposes functional groups (eg. -OH, -SH, -NH2, -COOH) allowing excretion or permitting compound to undergo phase II reactions
Examples of Phase 1 Reactions

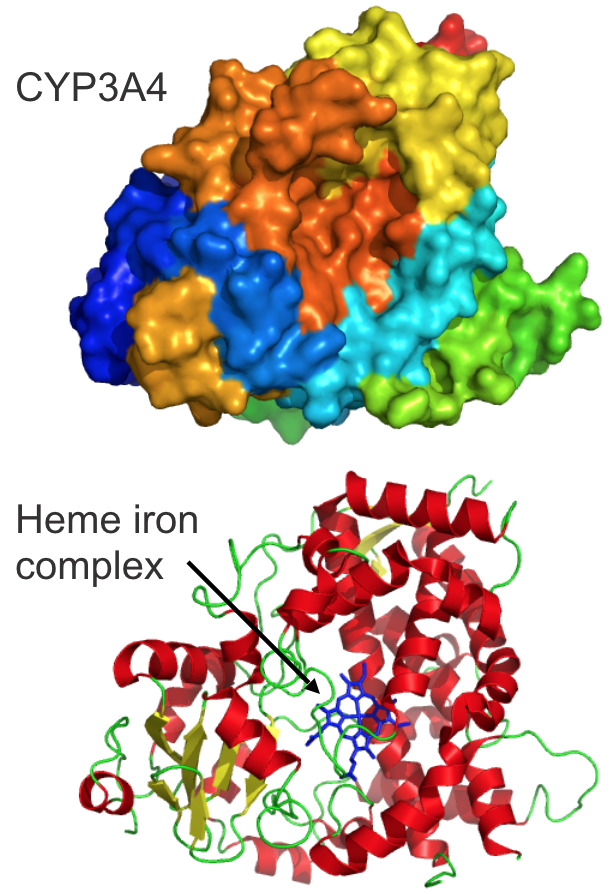
Cytochrome P450 structure
- P450 are heme-containing proteins.
- Polypeptide chains vary among CYPs and offer substrate specificity
- Basic reaction:
- Mono-oxygenation by one atom of oxygen into the substrate. The other oxygen atom is reduced to water
- Substrate (RH) + O2 + NADPH + H+ –––> Product (ROH) + H20 + NADP+
Activation of prodrugs
Bioactivation of prodrug (inactive) to active metabolite

Drug Metabolism: Phase 2
Drug metabolism: Phase 2
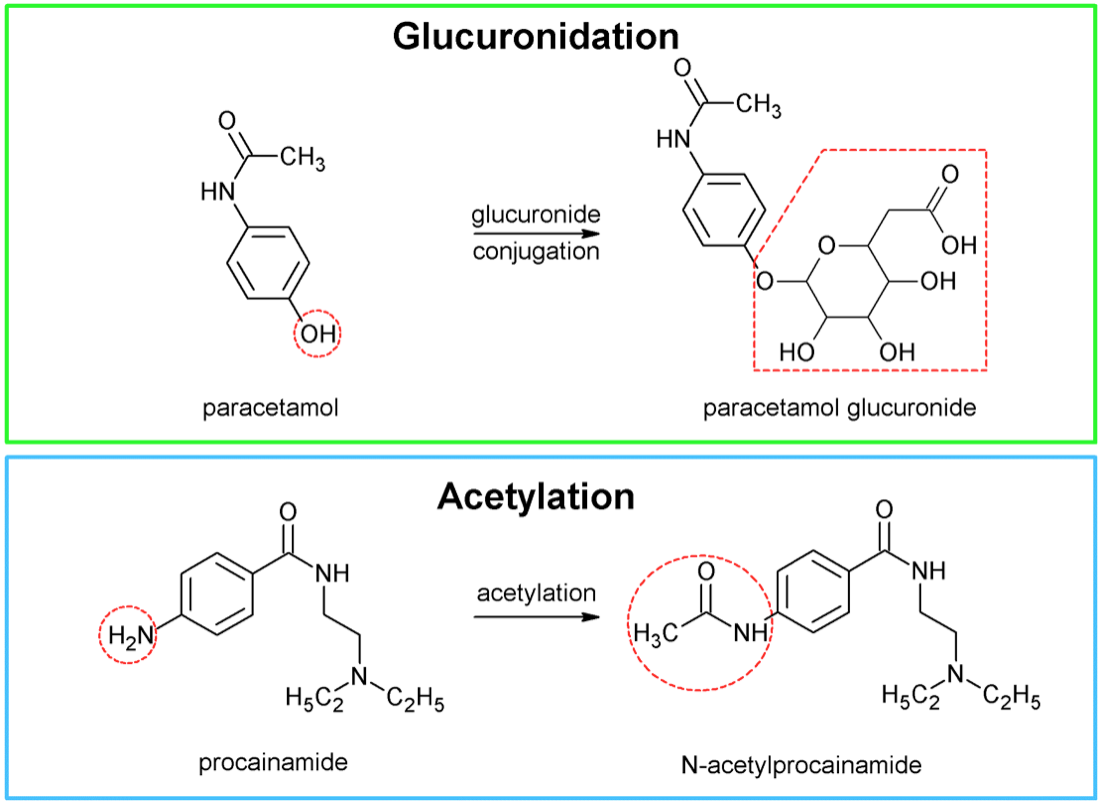
- Phase II reactions
- Biosynthetic reactions (require energy) where compound or phase I-derived metabolite is covalently linked to an endogenous molecule (conjugate)
- Conjugate = glucuronic acid, amino acids, glutathione, sulphate, methyl, or acetyl groups
- Conjugation (eg hydroxyl, thiol, amino group)
- Makes the drug less lipid soluble (highly polar) and more readily excreted in the urine and bile
Phase 2 reactions
Metabolism 3

Drug metabolism 8
- Pathways
- Phase 1 only
- Phase 2 only
- Phase 1 followed by Phase 2

Biotransformation in hepatocytes
- Most occurs in the liver via microsomal (smooth endoplasmic reticulum) and non-microsomal (mitochondria, soluble) enzymatic reactions
Codeine pharmacokinetics (Martindale)
- Codeine and its salts are absorbed from the gastrointestinal tract. Ingestion of codeine phosphate produces peak plasma codeine concentrations in about one hour.
- Codeine is metabolised by O– and N-demethylation in the liver to morphine, norcodeine, and other metabolites. Metabolism to morphine is mediated by the cytochrome P450 isoenzyme CYP2D6, which shows genetic polymorphism.
- Codeine and its metabolites are excreted almost entirely by the kidney, mainly as conjugates with glucuronic acid.
- The plasma half-life has been reported to be between 3 and 4 hours after an oral or intramuscular dose.
- Codeine crosses the placenta and is distributed into breast milk.
Drug excretion 1 (kidney)
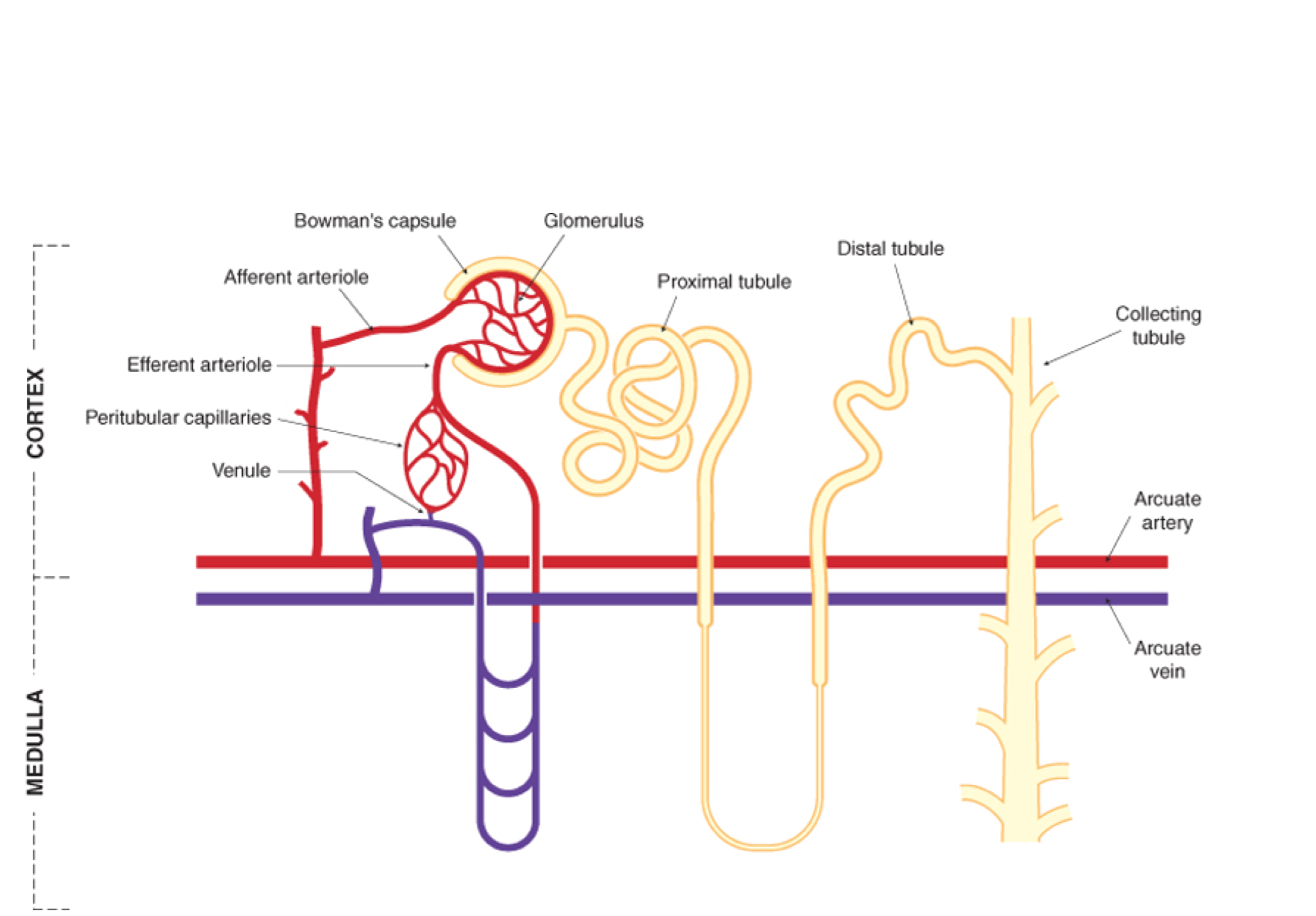



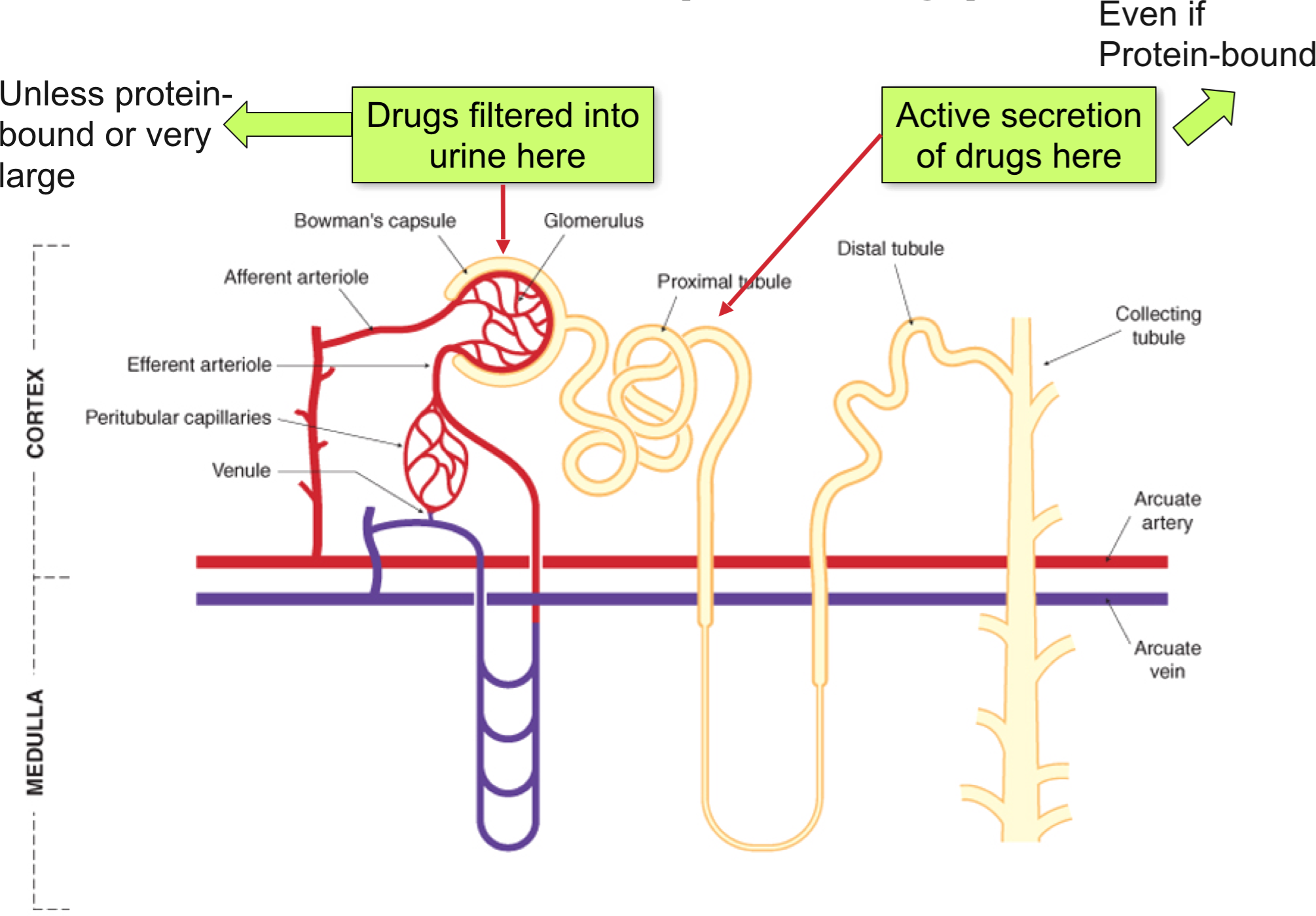


Renal Excretion
Renal excretion
- Nearly all drugs cross the glomerular filter freely
- They will be efficiently excreted (ie. remain in tubular fluid) unless they are lipid soluble and can be re-absorbed into the blood
- The key function of metabolism is to make the drug molecule less lipid soluble (more water soluble/more charged)
- Drugs excreted unchanged: digoxin gentamicin, methotrexate
Drug excretion 3
- Renal excretion
- Protein bound drugs and large molecules such as heparin (anticoagulant) are not filtered
- 80% of plasma is unfiltered and is present in peritubular capillaries of the proximal tubule
- Two non-selective (acid/base) carrier systems actively secrete weak acids (OAT) and bases (OCT) into the renal tubule, and thus they are more rapidly excreted
- Not restricted by plasma protein binding (highly efficient, so easily removed from binding site)
- Potential for competition between two drugs
- May be useful eg probenecid competes with penicillin for secretion, so prolonged penicillin t1/2
Drug excretion 4


Drug excretion 5

Drug excretion 6
- Ionised drugs (which are filtered or actively secreted in proximal tubule) undergo little reabsorption and are excreted
- Lipophilic drugs diffuse back (reabsorbed) into blood therefore not eliminated
- Drugs bound to plasma proteins are unable to be filtered but are subject to tubular secretion (eg. penicillin)
Drug excretion 7
- Most drugs undergo
- a) glomerular filtration
- b) partial tubular reabsorption
- Some only undergo
- c) tubular secretion
Biliary and Faecal Excretion
Biliary and faecal excretion
- Any unabsorbed orally administered drugs are excreted via faeces
- Low MW drugs (i.e. <325 in rats, <500-700 in man) are poorly excreted in bile
- Above this MW some compounds transferred to from plasma to bile (active transport system) then GIT then faeces in appreciable amounts (vercuronium)

Enterohepatic recirculation 2
- Bile acids are amphipathic (they have some water and some lipid solubiity) and allow absorption of fats, fat soluble vitamins etc.
- Bile is delivered to duodenum and 95% of bile acids are reabsorbed in ileum
- Portal vein delivers bile back to hepatocytes
- Hepatocytes extract bile acids efficiently
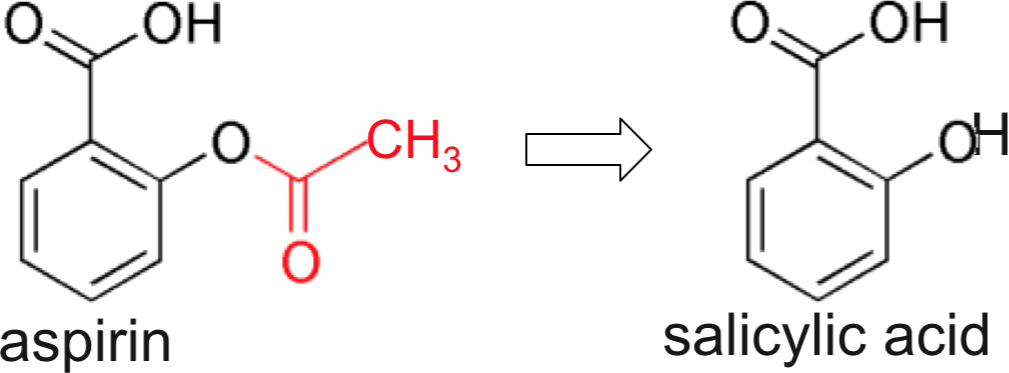
Enterohepatic recirculation 1
- β-glucuronidase from gut microflora removes glucuronide, reforming original drug that can then re-enter hepatic circulation
- Prolongs duration of action of affected drug
- Important for:
- morphine
- aspirin
- chloramphenicol (antibiotic)
- digoxin(inotropic agent)
Factors Affecting Excretion of Drugs
Factors affecting excretion of drugs
- High degree of ionisation
- High degree of water solubility
- Non-ionised / lipid soluble substances are reabsorbed and therefore not excreted
(Opposite to those required for absorption from GIT)


